
皮肤僵硬综合征一家系调查并文献复习
2020-06-06 00:58:24孟紫媛唐利利陈连军张学军
中国麻风皮肤病杂志 2020年6期
陈 敏 孟紫媛 唐利利 张 峥 陈连军 张学军
1安徽医科大学第一附属医院皮肤性病科,合肥,230022;2复旦大学附属华山医院皮肤性病科,上海,200040
皮肤僵硬综合征(stiff skin syndrome, SSS)是一种罕见的结缔组织病,由Esterly和McKusick[1]于1971年首次描述并命名。该病发病年龄较早,多发生于婴儿期或儿童期。其临床特点为皮肤增厚,如石头般坚硬,伴有轻度毛发增多,屈曲性关节挛缩活动受限,皮损常见于大腿和臀部。
SSS的病因和发病机制尚不清楚。Loeys等[2]提出SSS是由FBN1基因杂合突变引起的,该突变激活了转化生长因子β(TGF-β)结合蛋白样结构域4(TB4),扰乱了FBN1与细胞外基质之间的相互作用,从而引起真皮深层、皮下组织细胞、肌肉筋膜胶原沉积增多。我们对一SSS家系进行全基因组外显子测序(whole-exome sequencing)分析,但未找到其致病基因的突变位点,基因检测阴性,我们结合文献资料,总结SSS致病基因的特点,分析基因检测阴性的相关因素。
1 临床资料
先证者,男,11岁。因“右下肢皮肤变硬7年余,关节活动受限6年”就诊。患者3岁半时,家人无意间发现患者右大腿内侧皮肤变硬,后逐渐向右侧小腿、右侧臀部、腰部及腹部发展, 1年前右侧大腿、臀部僵硬部位出现毛发增多、增粗。病程中,患者无发热、关节肿痛、吞咽困难、雷诺现象。患者既往体健,否认外伤史,否认药物过敏史,对芒果过敏。患者父母无血缘关系,但患者曾祖父母是近亲结婚(姑表亲),患者家族中有两人(II2,IV2)(图1)与患者表现出相似的症状(图2)。
体格检查:一般情况良好,营养、发育及智力均正常。心肺系统未见明显异常,腹部膨隆,脊柱前屈,右膝关节活动轻度受限。皮肤科检查:腰腹部、臀部、右腿皮肤大片淡褐色硬斑,皮肤纹理正常,其上毳毛明显增多,表面凹凸不平,触之皮肤呈木板样硬,与皮下组织粘连,不能捏起(图2)。
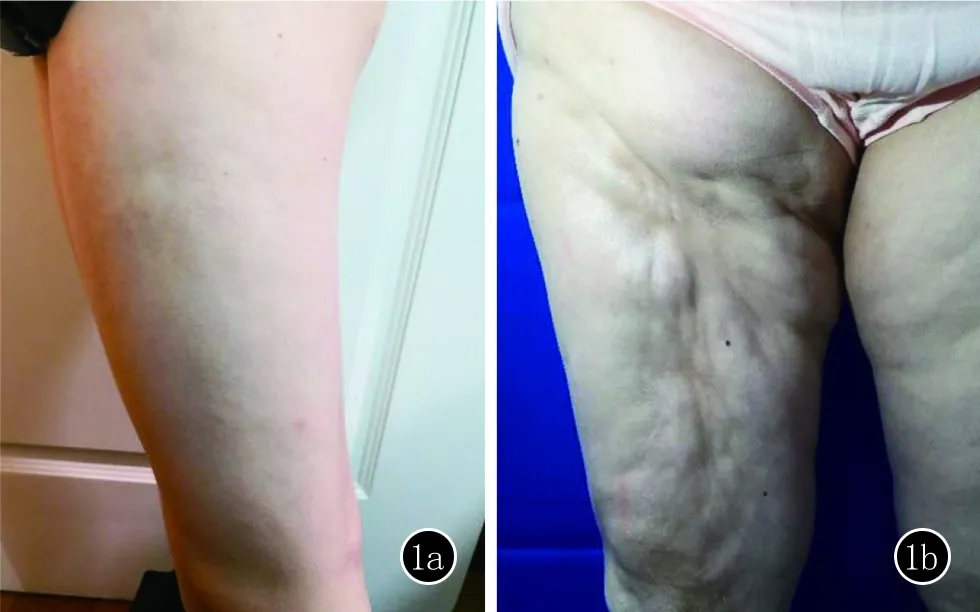
图1 1a、1b:II2,IV2患者临床图片
实验室及辅助检查:血常规、尿常规、肝肾功能、心肌酶酶谱、免疫球蛋白、补体C3、C4、血沉、肌电图,髋关节及双下肢X片无明显异常,抗ds-DNA抗体、抗核提取物(ENA)抗体(包含抗Scl-70抗体,抗Sm抗体,抗SSA、SSB等抗体)均阴性,抗核抗体(ANA)阳性,滴度(1∶100),颗粒型。取患者右侧大腿皮损行组织病理示:表皮轻度乳头瘤样生长,偶见基底细胞空泡变性,真皮网状层内胶原略增殖及纤维化,间杂有大小不一的脂肪团块,浅丛细血管周围小片状淋巴细胞浸润(图3a~3b)。阿新蓝染色示:真皮中下部部分区域胶原束间染色阳性反应(图3c)。诊断:皮肤僵硬综合征。目前治疗上主要以功能锻炼,理疗,改善局部微循环,预防关节挛缩和功能限制为主。
家系调查发现(图4),该家系4代共27人,其中3人患病,男2人,女1人,发病年龄分别为15岁(II2)、3.5岁(IV1)和3岁(IV2)。我们采集患者及其母亲的外周血进行了全基因组外显子测序,未发现既往报道致病基因FBN1基因的突变。
2 讨论
SSS是一种罕见的皮肤遗传疾病,多呈常染色体显性遗传[2,3]。SSS一般在婴儿期或在出生后6年内发病,但由于疾病进展缓慢,常较晚才能确诊。其特点为皮下组织细胞和肌筋膜的非炎症性纤维化,导致皮肤硬化,并干扰下位关节的运动,导致步态和姿势异常,病情严重者可致限制性胸廓通气功能障碍。该病最常见的受累部位是大腿、臀部和腰部,但在某些情况下近端手臂和肩胛带也会受累。实验室检查一般无明显异常,偶见IgA及IgG[4]免疫球蛋白降低的报道。本例患者ANA低滴度阳性,这在以往的报道中是没有发现的。虽然经典的病例描述不包括内脏受累,但某些病例可能伴有食管运动障碍、胃食管反流、腹痛、锁骨和远端指骨的进行性骨溶解[5],甚至有急腹症致死的报道[6]。

图2 2a~2c:先证者腰腹部、右侧臀部、大腿皮肤僵硬,腰部及右侧大腿皮肤僵硬部位毳毛轻度增多;2d:髋关节正位x线影像图

图3 3a:表皮大致正常,真皮网状层内胶原增生及纤维化,未见明显炎症细胞浸润(HE,×100);3b:胶原纤维组织间可见脂肪组织(HE,×200);3c:阿新蓝染色阳性(×200)
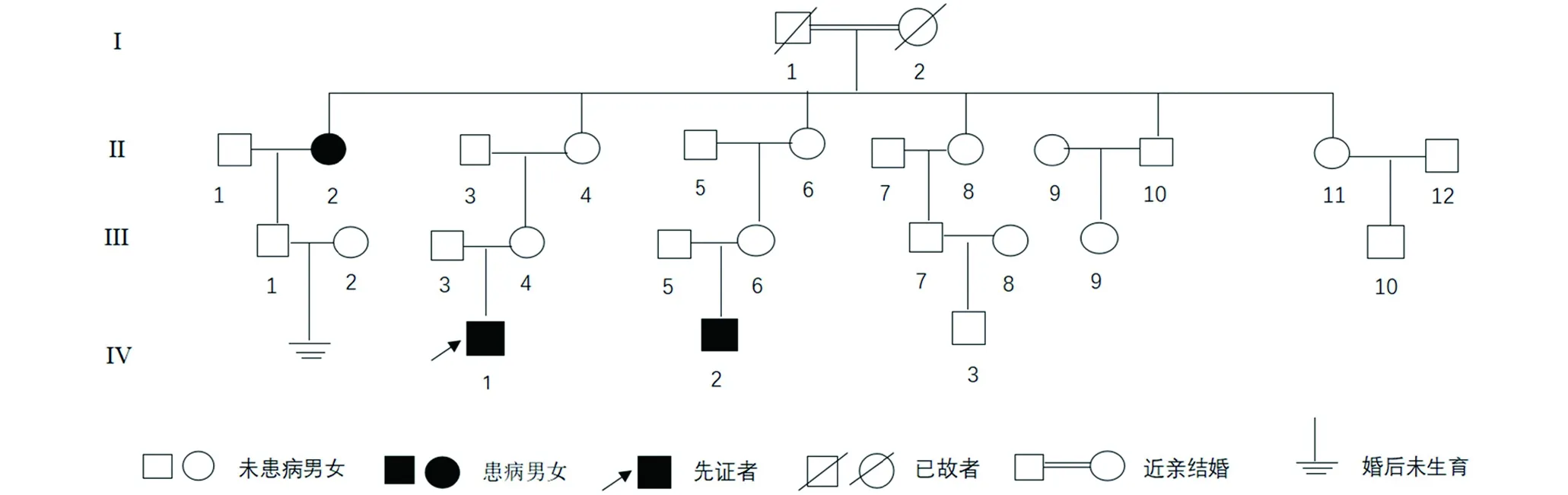
图4 家系图
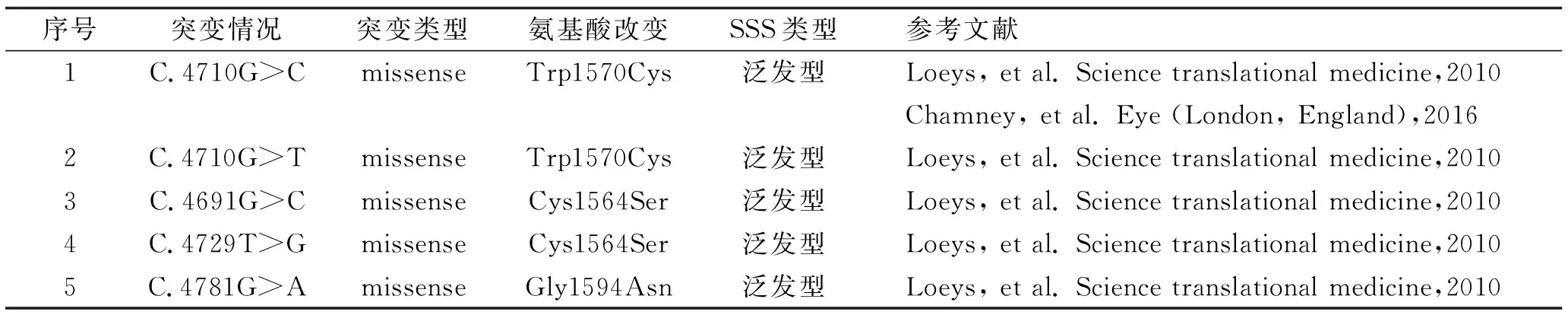
表1 SSS患者FBN1致病基因突变总结
SSS的病因及发病机制目前尚不清楚。目前主要认为FBN1基因的突变与其发病有关。目前关于SSS基因筛查的病例报道较少[2,3,7],迄今为止,仅发现FBN1基因杂合突变会导致SSS的发病,最常突变为外显子37处发现4170位碱基G突变成C或T(4710G>C或4710G>T),使得1570位氨基酸由色氨酸变成了半胱氨酸。在1例混合型SSS中,发现外显子38处发生错义突变(C.4781G>A),导致甘氨酸变成了天冬酰胺[2](表1)。值得注意的是,目前所报道的存在FBN1基因突变的家系均符合常染色体显性遗传特征,且受累患者均属于泛发型SSS。Maillet-Lebel等[7]曾报道过1例节段型SSS的散发病例,经基因检测未能在外周血DNA中发现FBN1基因突变,这与我们报道的相一致。我们推测泛发型SSS与节段型SSS可能存在不同的遗传机制。我们结合临床信息及测序试验,总结分析导致基因突变筛查阴性可能的原因:(1)全外显子测序技术仅能覆盖人类基因组近95%的编码区域,可能会丢失部分重要的遗传信息,导致基因检测结果出现假阴性[8];(2)由于WES技术的限制,可能无法检测到结构变异以及非编码区变异,而结构变异和非编码区变异也可能与疾病的发病相关[9];(3)WES是一种通过对目标区域捕获技术对基因组外显子区域富集的基因检测技术,这意味着在进行目标区域捕获时,可能存在捕获不均、捕获偏差的现象;(4)SSS可能存在其他的突变形式比如大片段重排,需要通过基因重排的检测技术进行甄别;(5)在我们的患者外周血DNA中没有发现致病基因,嵌合突变是可能的解释,然而不做皮损内组织测序是无法证实的。由此可见,皮肤僵硬综合征存在高度的遗传异质性,其发病机制还需进一步的研究。
Myers等[10]根据SSS患者的发病年龄、临床严重程度等,将SSS分为节段型SSS和泛发型SSS。泛发型SSS指皮损累及双侧伴关节活动受限,节段型SSS指皮损以单侧受累为主,可伴有关节活动受限,一般程度较轻。回顾分析52例SSS患者后发现[10],节段型SSS和泛发型SSS在发病年龄和临床严重程度方面存在显著差异。节段型SSS发病年龄较晚,平均4.1岁,以单侧受累为主,症状较轻,预后较好;而泛发型SSS平均发病年龄为1.6岁,通常累及双侧,患者常伴有不同程度的关节活动受限。我们对近年来国内外报道包括本例患者在内的61例SSS病例进行了回顾性分析,其中节段型SSS 22例,泛发型SSS 39例。在本组资料中,伴有关节活动受限有41例,关节活动受限部位最常见于膝关节、髋关节、肘关节等大关节(表2)。 SSS的组织病理学特征在节段型和泛发型之间并无明显差异。两者的特征都是胶原束增厚,非炎症性纤维化,结缔组织黏蛋白增加。在既往报道的病例中,并不是所有的病例都有筋膜累及,有些病例仅提示真皮轻度纤维化。故Jablonska等[11]把先天性筋膜发育不良归类为SSS的一种临床亚型。先天性筋膜发育不良的病理特征主要为筋膜增厚,且不伴有黏多糖的沉积。我们的患者病理表现未见筋膜的受累及炎症细胞的浸润,且阿新蓝染色阳性提示真皮胶原束间有粘蛋白样物质沉积,故不能将其归为先天性筋膜发育不良。本家系患者的皮损均以累及单侧为主,临床表现符合节段型SSS的特征。患者II2被发现患病时,症状已较为明显,故考虑实际发病年龄应远早于15岁。其中先证者临床症状最为严重,已有关节活动受限的表现。

表2 节段型SSS与泛发型SSS活动受限部位统计 例(%)
根据SSS患者典型的临床表现、皮损组织病理可确诊SSS,但仍需与硬皮病、结缔组织痣、平滑肌错构瘤等疾病相鉴别。局限性硬皮病可能有潜在的筋膜和肌肉受累,导致关节活动能力受损和肢体不对称,使其在临床上与SSS难以区分,但其组织病理表现为胶原均质化,无黏多糖沉积及明显的成纤维细胞增生。结缔组织痣通常局限于真皮,常伴有弹性纤维分布异常,而SSS仅涉及皮下及筋膜下,并不涉及真皮层,两者较易区分[12]。平滑肌错构瘤组织病理可见真皮中增多的平滑肌纤维。
目前针对SSS尚无有效的治疗手段。口服和外用糖皮质激素、甲氨蝶呤、补骨脂素以及PUVA治疗效果都较差[13]。国外有文献报道吗替麦考酚酯[14]、氯沙坦[7]对节段型SSS治疗有效。局部注射糖皮质激素可改善临床症状[15],但本病属于非炎症纤维化性疾病[16],故不推荐使用,以免引起过多的不良反应。国内有文献报道皮损内局部注射透明质酸酶配合热敷及外涂肝素钠乳膏对皮损有一定的软化作用[17]。由于这种疾病的治疗方法有限,及时诊断并及早物理治疗对预防关节活动受限以及维持生活质量至关重要。
猜你喜欢
西南国防医药(2016年7期)2016-12-01 06:01:16
米娜·女性大世界(2016年8期)2016-08-17 17:15:15
Coco薇(2016年5期)2016-06-03 08:53:30
中国卫生标准管理(2015年15期)2016-01-15 02:58:42
爆笑show(2015年9期)2015-10-24 15:03:28
实用手外科杂志(2015年3期)2015-08-27 01:53:18
中国医疗美容(2015年4期)2015-04-27 02:24:09
西部皮革(2015年3期)2015-04-16 05:07:31
中华皮肤科杂志(2014年3期)2014-12-19 12:54:41
食品科学(2013年13期)2013-03-11 18:24:42
