
金属有机骨架-分子印迹复合材料的制备、表征及其对吗啉的吸附性能*
2020-06-05 10:34龚梦婷宋俊杰田海希张朝晖
功能材料 2020年5期
龚梦婷, 宋俊杰,田海希,李 辉 ,2,张朝晖
(1. 吉首大学 化学化工学院,湖南 吉首, 416000;2.吉首大学 植物资源保护与利用湖南省高校重点实验室,湖南 吉首, 416000)
0 引 言
分子印迹聚合物是一种依据目标分子大小、形状及功能基团而设计构造的新型材料,由于这种材料与目标分子具有多重匹配性,将其用于对目标化合物进行固相萃取分离时,具有类似于抗体识别的高选择性能[1-2]。迄今,分子印迹材料已广泛用于固相萃取、色谱分离、化学传感、药物传输及分子催化等领域[3-6]。分子印迹材料尤其适用于复杂混合物中微量及痕量组分的选择富集和分离分析[7]。表面印迹技术是获取分子印迹材料的重要手段之一,采用该种方法获得的印迹材料,活性位点广布于材料表面,在萃取的吸附-脱附过程中具有较快的传质动力学,可大大提高分子印迹固相萃取效率[8-10]。与传统的载体(如硅胶、氧化铝、碳纳米管等)相比,金属有机骨架具备比表面非常高、吸附能力强等特点[11-12],但其对目标化合物缺乏预定选择性,不适宜对特定分子的选择富集。将具有高选择性的印迹材料接枝到金属有机骨架表面,将分子印迹聚合物的高选择吸附能力与金属有机骨架的高吸附容量有机结合,可大大提高所获新材料对特定目标分子的吸附效能。
吗啉是一种含氮杂环化合物,常用于制备水果表皮被膜剂(如果蜡等)[13],将其涂覆于新鲜水果(如苹果、橙柑等)表面,具有抑制水果水分挥发、减少表皮皱缩、增强水果视感等效果[14-15]。我国《食品添加剂使用标准》法规允许适量食用。但这种被膜剂因长时间受空气及光照影响,易发生分解释放出吗啉,残留于水果表皮甚至渗透进果肉中,从而对食用者产生潜在毒害作用[16-17]。探寻灵敏准确快速的吗啉检测方法具有积极意义。国内外科研工作者在食品中吗啉的检测方面做出了一些有价值的研究和探索。张博及陈达炜等人构建了分散固相微萃取-同位素内标法-超高效液相色谱-串联质谱法测定水果中吗啉残留[18-19]。沈葹等建立了固相萃取-同位素稀释高分辨质谱法检测苹果和橙柑类水果中吗啉含量[20]。Luong等采用气相色谱法对超痕量吗啉进行分析[21]。Joseph等测定了食品包装纸中吗啉含量,显示吗啉可从水果表层转移到包装材料[22]。Hengel等使用酸化甲醇提取高效液相色谱-质谱-质谱联用技术检测水果中残留的吗啉[23]。对水果中残留吗啉进行分析时,为了提高分析准确性,常需对样品中的吗啉进行富集和浓缩。不同类型的离子交换树脂、改性硅胶等材料常用于对吗啉的固相萃取[24-26],但由于这些材料对目标化合物的选择性不够高,实际应用中富集分离效果受限。
本文拟以金属有机骨架MIL-101为基质,用乙二胺对其进行表面化学修饰后,再接枝吗啉印迹聚合物制备出金属有机骨架-分子印迹聚合物新型复合材料,并以之为吸附剂,探讨其对吗啉的吸附性能。
1 材料与方法
1.1 材料与试剂
九水硝酸铬(AR)、对苯二甲酸(AR)及乙二胺(AR)均购自天津市光复精细化工研究所;氢氟酸(AR)购自上海申博化工有限公司;N,N-二甲基甲酰胺(AR)和甲苯(AR)购自成都金山化工试剂有限公司;甲醇(AR)、乙醇(AR)、四氢呋喃(AR)、醋酸(AR)、Na2HPO4(AR)和NaH2PO4(AR)均购自天津永大化学试剂有限公司。甲基丙烯酸(AR)、二乙烯基苯(AR)和偶氮二异丁腈(AR)均购自美国阿拉丁工业公司。标准物质吗啉、甲基吗啉、乙基吗啉和甲基吗啉氧化物均购自成都康邦生物科技有限公司。
1.2 仪器与设备
LC-20A 型高效液相色谱( 日本岛津公司)用于定性和定量分析; IR-Affinity-1型傅立叶变换红外光谱仪(日本岛津公司)用于红外光谱分析; S-3400型扫描电子显微镜(日立公司)用于观察合成物质的颗粒大小及形貌特征;KQ-250E 型超声波清洗器( 昆山市超声仪器有限公司); UV-2550 型紫外可见分光光度计(日本岛津公司) ;BZF-50 型真空干燥箱(上海博迅实业有限公司);FA2104N 型电子分析天平(上海民桥精密科学仪器限公司);TGL-16型高速台式离心机( 江苏金坛市大地自动化仪器厂);FA2104N型电子天平(上海菁海仪器有限公司);100mL水热反应釜(中国临朐广利机械设备有限公司)。
1.3 方法
1.3.1 复合材料的制备
金属有机骨架-分子印迹聚合物复合材料的制备过程如图 1 所示,主要包括三步:(1)金属有机骨架 MIL-101 的制备;(2)MIL-101 氨基化修饰制备ED-MIL-101;(3)表面接枝吗啉印迹聚合物 。
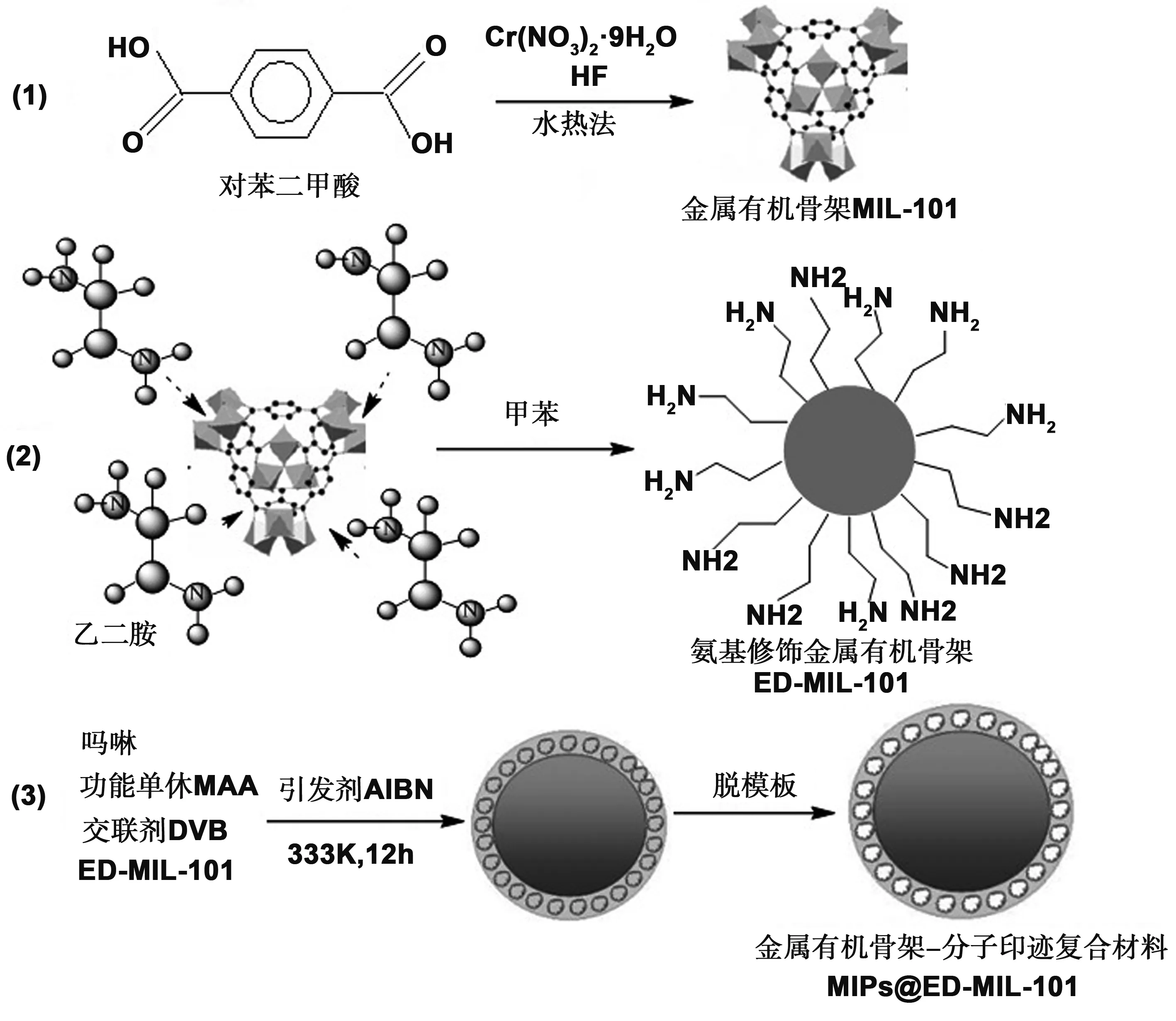
图1 金属有机骨架表面印迹聚合物的制备流程Fig 1 Preparation process chart for the MOFs-MIPs composite material (MIPs@ED-MIL-101)
1.3.2 MIL-101 的制备
按文献方法[11]制备金属有机骨架MIL-101。制备过程为:分别称取0.01 mol九水硝酸铬及0.01 mol对苯二甲酸,加入60 mL去离子水和0.2 mL氢氟酸,超声搅拌30 min后,将混合液倒入100 mL水热反应釜中,密封,置于493 K恒温干燥箱中晶化8小时,取出并冷却至室温后,移至塑料离心管中离心分离,吸出上清液。用去离子水重复洗涤固体3次,吸出上清液,用 20 mLN,N-二甲基甲酰胺(DMF)洗涤3次,吸出上清液。再将固体转移至水热反应釜内,加入50 mL乙醇,并置于373 K恒温干燥箱中反应20 h。取出反应混合物并冷却至室温,过滤,用20 mL乙醇重复洗涤固体3次。所得绿色粉末在423 K温度下真空干燥24 h,研磨过筛,即得金属有机骨架材料MIL-101。
1.3.3 MIL-101的氨基化修饰
称取4.0 g MIL-101 固体粉末,装入250 mL烧瓶中,加入100 mL无水甲苯,搅拌均匀后加入5.0 mmol乙二胺,回流12h。冷却,过滤,用 50 mL无水乙醇反复洗涤固体3次,固体真空干燥24h,研磨,所得灰色粉末即为氨基化修饰的金属有机骨架 ED-MIL-101。
1.3.4 复合材料(MIPs@ED-MIL-101)的制备
取0.5 mmol吗啉、2.0 mmol甲基丙烯酸(MAA)、5.0 mmol二乙烯基苯(DVB)及0.02488 g偶氮二异丁氰(AIBN)溶于3.0 mL体积比2∶1的甲苯-四氢呋喃混合溶液中,加入0.4 g ED-MIL-101,超声混合5 min,通氮气5 min,得到的混合溶液置于60 ℃水浴中反应12 h。将生成的固体研磨过筛,所得粉末用20.0 mL体积比9∶1的甲醇-乙酸混合溶液洗涤多次,直到洗涤液中检测不到吗啉为止,过滤,固体再用20.0 mL去离子水反复洗涤3次后,过滤,所得固体于333 K温度下真空干燥12 h。即得MIPs@MIL-101印迹聚合物。非印迹复合材料(NIPs@ED-MIL-101)的制备步骤与印迹聚合物相同,但在制备时不加模板分子。
1.3.5 吸附动力学
在20 mL含 0.5 mg mL-1吗啉的甲醇-乙腈(2∶1,V/V)溶液中,加入40 mg印迹复合材料, 振摇。每隔0.5 h,取少量上清液,用高效液相色谱法测定其中吗啉浓度,按(1)式计算不同时间下分子印迹聚合物对吗啉的吸附量。
1.3.6 等温吸附
将10.0 mg印迹复合材料分别加入到5 mL 含有 0.2, 0.4, 0.6, 0.8, 1.0, 1.2, 1.4, 1.6, 1.8 mg/mL吗啉的甲醇-乙腈(2∶1,V/V)混合溶液中,在298 K下,静态吸附3 h后,过滤,用高效液相色谱法测试滤液中吗啉的浓度,按(1)式并计算不同底物浓度下的平衡吸附量。
1.3.7 选择性测试
分别在10 mL、0.5 mg mL-1的吗啉、甲基吗啉、乙基吗啉和甲基吗啉氧化物的甲醇-乙腈溶液(2∶1,V/V)中,加入10.0 mg印迹复合材料,体系置于298 K水浴中,吸附3h后,液相色谱法测定各溶液中化合物浓度,并按(1)式计算各物质的吸附量(q, mg/g)和分布系数(kd)及选择因子(k)等参量
(1)
Kd=q/ρe
(2)
K=Kd(Morpholine)/Kd(Related compounds)
(3)
式中,ρ0(mg/mL)为底物初始浓度,ρe(mg/mL)为底物平衡浓度,V(mL)为溶液体积,m(g)为聚合物。
1.3.8 水果粗提物溶液的制备
冰糖橙购自湘西农贸市场。 将冰糖橙用研钵磨成浆,称取匀浆样品200.0 g于烧杯中,加入150.0 mL体积比为2∶1的甲醇-乙腈溶液,置于超声介质中超声处理20 min,离心分离,液体通过减压蒸馏至糊状,用少量甲醇溶解后,得到水果粗提物溶液,备用。
1.3.9 高效液相色谱分析
高效液相色谱分析以C18 柱(4. 6 mm × 250 mm,5 μm) 为分离柱,流动相为甲醇-0.002 mol·L-1pH 6.5 磷酸缓冲液含0.5%的三乙胺(体积比60∶40),流速1.0 mL /min,检测波长250 nm,进样体积10 μL。采用标准曲线法进行定性和定量分析。
2 结果与分析
2.1 金属有机骨架-分子印迹复合材料的制备及表征
本文采用水热合成法首先制备金属有机骨架材料MIL-101,再对其进行化学修饰,然后采用表面接枝技术制备金属有机骨架-吗啉印迹复合材料,整个制备过程分为三步,如图1所示。由于MIL-101(Cr)的中心金属铬上存有大量不饱和活性金属位点,能与富电子官能团(如胺基、巯基、叠氮基等)发生螯合作用,得到功能化的MIL-101。我们采用乙二胺对金属有机骨架进行功能化修饰,便于后续分子印迹聚合物的表面接枝。在分子印迹复合材料的制备过程中,分别得到了金属有机骨架材料MIL-101、氨基化修饰的金属有机骨架材料ED-MIL-101、分子印迹复合材料MIPs@ED-MIL-101及非印迹复合材料NIPs@ED-MIL-101。
4种材料的扫描电镜图如图2所示。从中可以看出,MIL-101及ED-MIL-101晶体颗粒大小约为2~5μm,当ED-MIL-101材料表面接枝分子印迹及非印迹聚合物后,晶体颗粒变大,但聚合物颗粒形状仍无明显规则。显示了金属有机骨架表面的成功接枝。
图3给出了金属有机骨架MIL-101、ED-MIL-101、吗啉印迹复合材料MIPs@ED-MIL-101及非印迹复合材料NIPs@ED-MIL-101的红外吸收光谱 。 在MIL-101的红外光谱中 ,3 102 cm-1处为对苯二甲酸分子中O-H键的伸缩振动吸收峰,1 682 cm-1处的吸收为分子中残余C=O键的伸缩振动吸收峰,1 490 cm-1处为骨架中-(O-C-O)-的伸缩振动峰,1 143 cm-1处的吸收峰是苯环中C-N键的伸缩振动吸收峰;在ED-MIL-101的红外光谱中,3 624 cm-1处吸收峰是乙二胺分子上-N-H 的伸缩振动吸收峰,3 105 cm-1处为对苯二甲酸分子中O-H键的伸缩振动吸收峰,1 675 cm-1处为C=O键的伸缩振动吸收峰,1 491 cm-1处为骨架中-(O-C-O)-的伸缩振动峰,在1 144 cm-1处的吸收峰是C-N伸缩振动吸收峰,820 cm-1处的吸收峰是-NH的变形振动吸收;在分子印迹复合材料(MIPs@ED-MIL-101)的红外光谱中,3 617 cm-1处吸收峰是分子中-N-H的伸缩振动吸收峰,3 125 cm-1处为分子中O-H键的伸缩振动吸收峰,1680cm-1 处为C=O键的伸缩振动吸收峰,1 490 cm-1处为分子中-(O-C-O)-的伸缩振动峰,在1 162 cm-1处的吸收峰是C-N伸缩振动吸收峰;在非印迹复合材料(NIPs@ED-MIL-101)的红外光谱中,3 627 cm-1处吸收峰是分子中-N-H键的伸缩振动吸收峰,3 107 cm-1处为分子中O-H键的伸缩振动吸收峰,1 685 cm-1处为C=O键的伸缩振动吸收峰,1 483 cm-1处为分子中-(O-C-O)-的伸缩振动峰,在1 150 cm-1处的吸收峰是C-N伸缩振动吸收峰。

图2 金属有机骨架(MIL-101)、氨基修饰金属有机骨架(ED-MIL-101)、印迹复合材料(MIPs@ED-MIL-101)及非印迹复合材料(NIPs@ED-MIL-101)的扫描电镜图Fig 2 SEM pictures of MIL-101, ED-MIL-101, MIPs@ED-MIL-101 and NIPs@ED-MIL-101
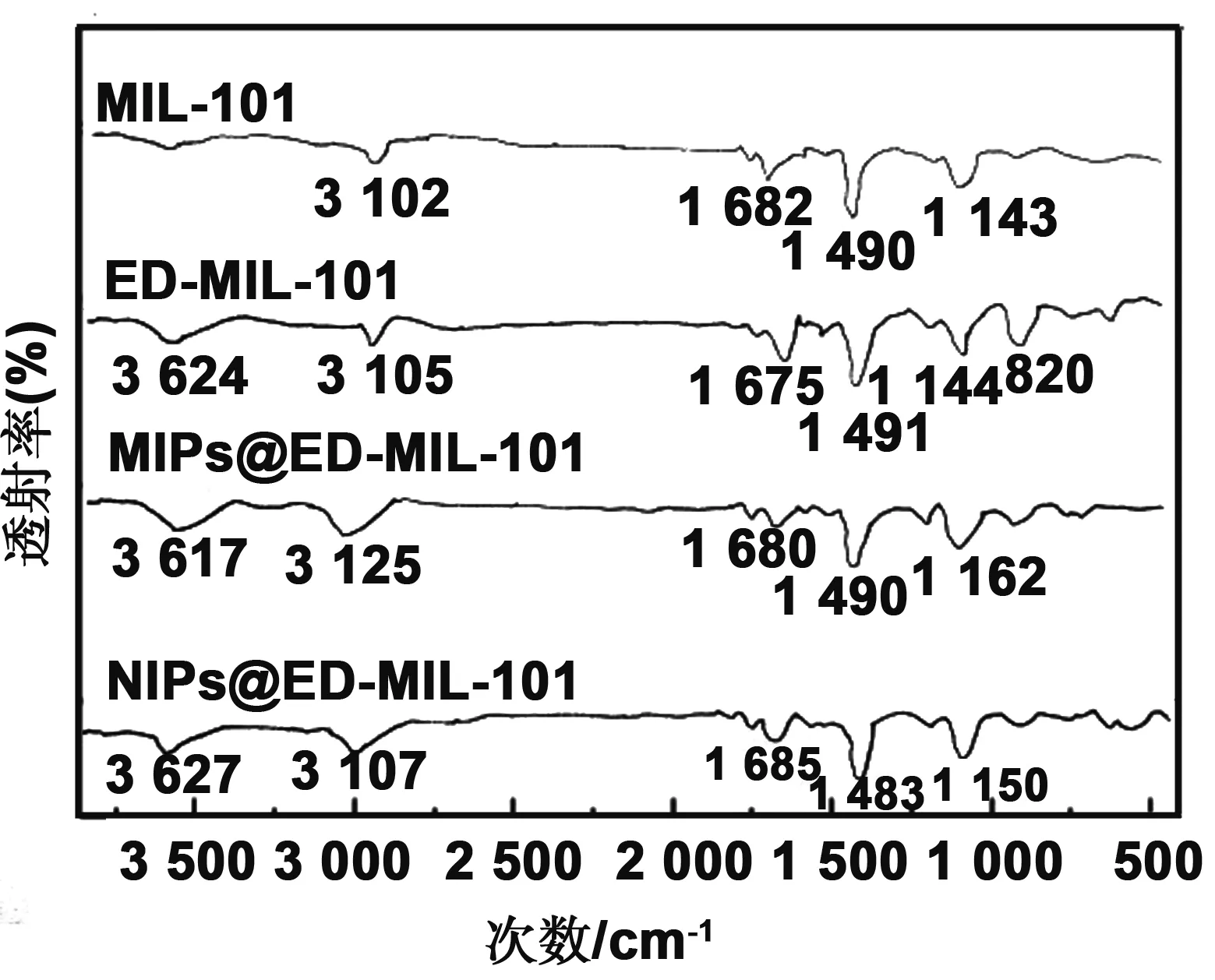
图3 金属有机骨架(MIL-101)、氨基修饰金属有机骨架(ED-MIL-101)、表面印迹聚合物(MIPs@ED-MIL-101)及非印迹聚合物(NIPs@ED-MIL-101)的红外光谱Fig 3 IR spectrum of MIL-101, ED-MIL-101, MIPs@ED-MIL-101 and NIPs@ED-MIL-101
2.2 分子印迹复合材料的吸附性能
2.2.1 吸附动力学
图 4 给出了分子印迹复合材料MIPs@ED-MIL-101的吸附动力学曲线。从中可以看出,分子印迹复合材料对模板的吸附量随时间增加而增大,当吸附时间为150 min时,达到吸附平衡,显示了较快的吸附动力学,这是由于印迹位点分布于材料表面的原故。

图4 分子印迹复合材料MIPs@ED-MIL-101对模板分子的吸附动力学曲线Fig 4 Adsorption kinetic curve for the template on the MIPs@ED-MIL-101
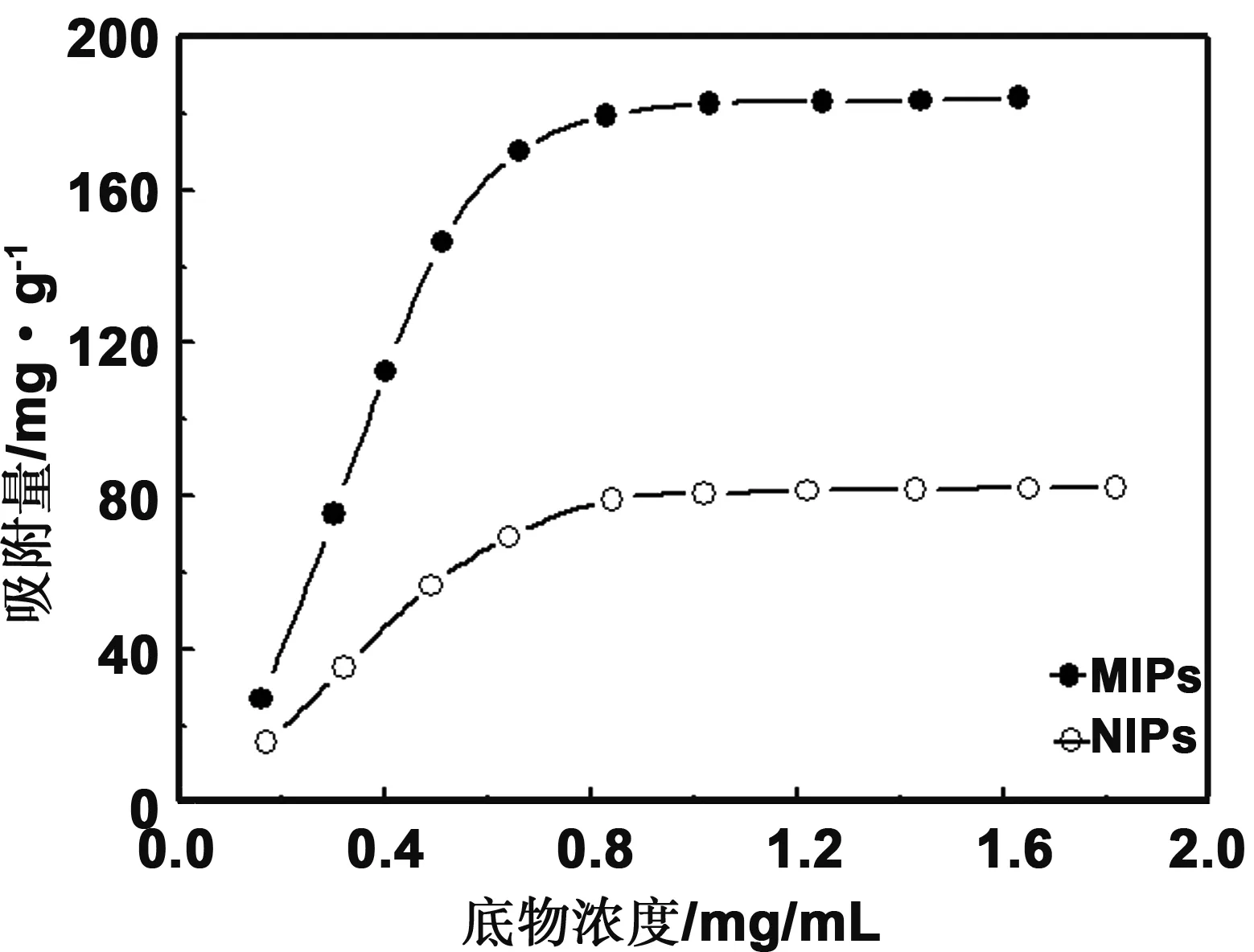
图5 298K时,分子印迹复合材料对模板的吸附等温线Fig 5 Adsorption isotherms for the polymer MIPs@ED-MIL-101 toward the template at 298 K
2.2.2 等温吸附
图5为298K下分子印迹及非印迹复合材料对模板分子的吸附等温线,可以看出当底物浓度增加时,二种聚合物材料对吗啉的吸附量增加;当底物浓度相同时,印迹复合材料的平衡吸附量高于非印迹复合材料,二者的饱和吸附量分别为183.3和 78.7 mg/g。印迹复合物的吸附能力高于非印迹复合物,主要是由于印迹复合材料基体中存有大量与模板分子大小、形状及结构相匹配的结合位点。
为了探讨印迹复合材料表面吸附位点的特征,采用Scatchard分析法对吸附等温线进行线性拟合,发现分子印迹复合材料表面主要存在两类吸附位点(如图6(a)所示),对高亲和位点,平衡离解常数K和最大表观吸附量Qmax分别为0.5679 g/L和326.5 mg/g;对低亲和位点,平衡离解常数K和最大表观吸附量Qmax分别为2.493 g/L和562.9 mg/g。这两类位点,一类源于模板分子的印迹作用,另一类来源于功能单体的随机分布。而非印迹复合材料表面主要存在一类吸附位点(如图6(b)所示),其平衡离解常数K和最大表观吸附量Qmax分别为1.036 g/L和120.4 mg/g。

图6 分子印迹及非印迹复合材料对吗啉吸附等温线的Scatchard分析Fig 6 Scatchard analysis of adsorption isotherms for MP on the MIPs@ED-MIL-101and NIPs@ED-MIL-101
2.2.3 分子印迹复合材料的吸附选择性
以吗啉及其结构相似化合物甲基吗啉、乙基吗啉、甲基吗啉氧化物为对象(其分子结构如图7所示),测试了分子印迹复合材料MIPs@ED-MIL-101、非印迹复合材料NIPs@ED-MIL-101及金属有机骨架MIL-101对吗啉分子的吸附选择性。表1分别给出了三种材料对四种化合物的分布系数(Kd)及其对模板的选择因子(k)。可以看出:金属有机骨架-分子印迹复合材料对模板的选择因子相对于甲基吗啉、乙基吗啉和甲基吗啉氧化物分别为2.07、2.48和2.24,均高于非印迹复合材料和MIL-101对模板分子的选择性。这是由于吗啉印迹位点负载于金属有机骨架MIL-101上,增加了所得新材料对模板分子的选择识别性能。
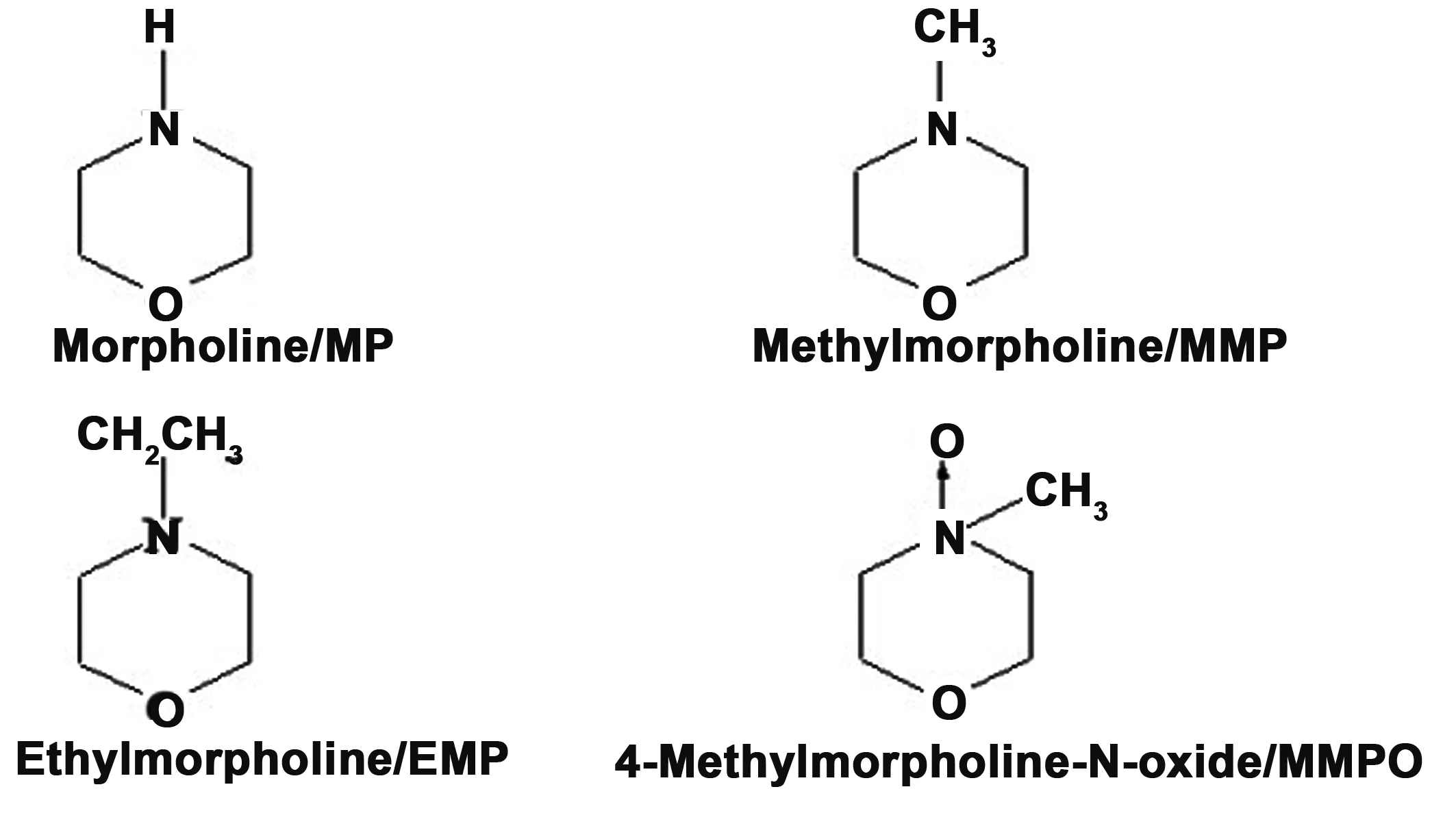
图7 几种化合物的分子结构Fig 7 Molecular structure of several compounds

表1 分子印迹复合材料、非印迹复合材料及金属有机骨架MIL-101对模板的吸附选择性
2.3 分子印迹复合材料固相萃取
2.3.1 模拟样品溶液的装载和洗脱
为考察分子印迹复合材料的固相萃取性能,首先测试了分子印迹复合材料萃取柱的装载容量。将0.15 g复合材料装入固相萃取器中,先用乙腈洗涤聚合物中的非选择吸附,再用甲醇洗涤其中的残余化合物并用乙腈平衡后进行装载容量的测试。将含5.0 mg·mL-1吗啉的甲醇溶液注入到固相萃取器上口,流出液中的吗啉浓度用紫外检测器进行在线检测。图8给出了吗啉在分子印迹及非印迹复合材料萃取柱上的突破曲线。可以看到:吗啉在分子印迹复合材料柱上的突破点(突破时间约为22 min)大于非印迹复合材料柱(突破时间约为12 min),分子印迹复合材料柱上的流出曲线平衡时间约为150 min,远高于后者(流出曲线平衡时间约为70 min),表明分子印迹聚合物的装载量高于非印迹聚合物柱。通过测定所收集流出液中的吗啉浓度,可知分子印迹及非印迹复合材料柱上吗啉的装载量分别为17.73和8.19 mg。

图8 吗啉在分子印迹及非印迹复合材料柱上的突破曲线Fig 8 Breakthrough curve for BP on the SPE cartridges filled with the MIPs@ED-MIL-101and NIPs@ED-MIL-101 as adsorbents, respectively
在固相萃取过程中,为了优化洗涤和洗脱过程,测试了8种萃取方法及各种方法中洗涤剂和洗脱剂的组成,如表2所示。每种方法涉及到三个洗涤步骤和1个洗脱步骤。表3给出了每种方法中各洗涤和洗脱步骤中吗啉的回收率。可以发现,在分子印迹复合材料柱的固相萃取过程中,当采用方法1#、2#和3#时,在Washing-1步骤中就有目标物流出,这显然不利于吗啉的富集;当采用方法4#、5#、6#和7#时,虽然在Washing-1步骤中没有检测出目标化合物,但在Washing-2和Washing-3步骤中,因有较多的目标物流失致使在Elution步骤中吗啉回收率不够高;最佳的萃取过程应采用方法8#,该种方法全部洗涤步骤中吗啉流失率最低(仅为14.81%),洗脱步骤吗啉回收率最高,达84.79,总回收率达99.6%,显示了较好的萃取效果。而非印迹复合材料柱不具有选择萃取效能。

表2 固相萃取吗啉的洗涤和洗脱过程×
*A: H2O; B: 甲醇; C: 乙腈; D:乙酸; E: 0.002 mol L-1Na2HPO4-NaH2PO4缓冲液
表3分子印迹复合材料固相萃取各洗涤和洗脱步骤中吗啉回收率
Table3RecoveryofMPobtainedbyusingMOFs-MIPscompositemaerialSPEineachstepofwashingandelutionprocedure
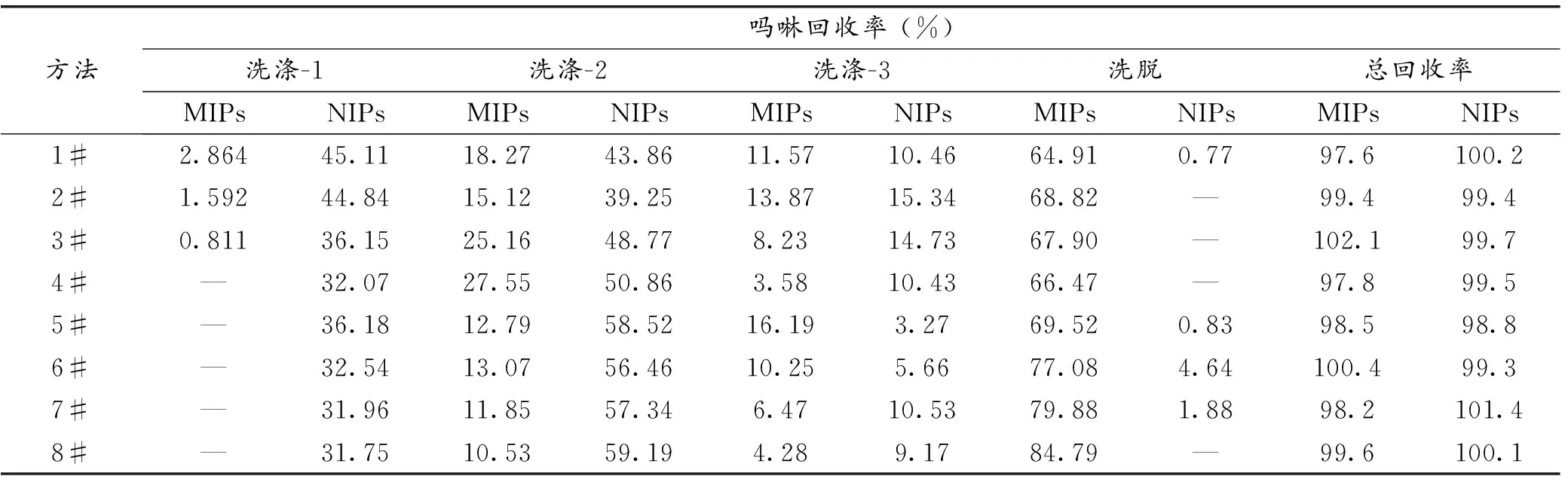
方法吗啉回收率 (%)洗涤-1洗涤-2洗涤-3洗脱总回收率MIPsNIPsMIPsNIPsMIPsNIPsMIPsNIPsMIPsNIPs1#2.86445.1118.2743.8611.5710.4664.910.7797.6100.22#1.59244.8415.1239.2513.8715.3468.82—99.499.43#0.81136.1525.1648.778.2314.7367.90—102.199.74#—32.0727.5550.863.5810.4366.47—97.899.55#—36.1812.7958.5216.193.2769.520.8398.598.86#—32.5413.0756.4610.255.6677.084.64100.499.37#—31.9611.8557.346.4710.5379.881.8898.2101.48#—31.7510.5359.194.289.1784.79—99.6100.1
—未检测出。

图9 水果粗提物(a)、分子印迹复合材料固相萃取洗涤液(b)和洗脱液(c)及非印迹复合材料固相萃取洗涤液(d)和洗脱液(e)的高效液相色谱图Fig 9 HPLC analysis of crude extract of fruit, effluent collected in the washing and elution steps for the MOFs-MIPs composite material SPE, and that in the washing and elution steps for the MOFs-NIPs composite material SPE, respectively
2.3.2 水果粗提物中吗啉的固相萃取
采用优化的方法8#执行对水果粗提物中吗啉进行富集和萃取分离。取20 mL水果粗提物的样品溶液注入到固相萃取柱上口进行装载,通过真空抽吸,使样品溶液快速流过萃取柱,收集流出液,高效液相色谱法测定所收集流出液中吗啉浓度,计算装载量。按照方法8#进行洗涤和洗脱。收集洗涤和洗脱步骤中的流出液,采用高效液相色谱法测定其中吗啉浓度,图9(a~e)分别给出了水果粗提物样品溶液、分子印迹复合材料柱及非印复合材料柱固相萃取过程中所收集洗涤液和洗脱液的高效液相色谱图。从图9(c)可以看出,水果粗提物经分子印迹复合材料柱富集和洗脱后,所收集流出液的色谱图中产物峰较为单一,仅见少量杂质峰,体现了印迹复合材料较高的富集和分离效能。而非印迹复合材料不能有效富集和分离目标化合物(图9d~e)。表4给出了分子印迹及非印迹复合材料固相萃取过程中各步骤吗啉的回收率,结果显示分子印迹复合材料可使水果粗提物中吗啉得到高效富集和分离,其单步洗脱中吗啉回收率高达80%以上。
表4分子印迹复合材料固相萃取水果粗提物中的吗啉
Table4MOFs-MIPscompositematerialsolidphaseextractionofMPfromsamplesolutionoffruitcrudeextract

步骤吗啉回收率 (%)分子印迹复合材料非印迹复合材料洗涤-11.5740.28洗涤-28.2350.39洗涤-310.068.82洗脱82.442.01总回收率102.3101.5
2.3 重现性测试
测试了金属有机骨架-分子印迹复合材料重复多次使用后对目标化合物吗啉的吸附性能。在10 mL含0.2 mg/mL吗啉的甲醇溶液中,加入20 mg分子印迹聚合物,吸附平衡后过滤,高效液相色谱法测定滤液中吗啉浓度,计算吸附量。然后用甲醇-冰醋酸(9∶1,V/V)混合溶剂洗净吸附在分子印迹复合材料中的吗啉,过滤并干燥后,复合材料再加入到10 mL 0.2 mg/mL的吗啉甲醇溶液中,吸附平衡后测定吸附量。按照相同的步骤重复测定9次,分子印迹复合材料的吸附量如图10所示。可以看出该材料重复使用多次后,吸附量变化不大,显示了较高的可重复使用性。

图10 分子印迹复合材料使用重现性测试Fig 10 Test of reusability for the MIPs@ED-MIL-101
3 结 论
以金属有机骨架MIL-101为载体,经表面氨基化修饰后,采用表面印迹技术成功接枝了吗啉印迹聚合物。这种新型印迹复合材料对模板具有较快的吸附动力学,在298 K时,对吗啉的饱和吸附量达183.3 mg/g,远高于非印迹复合材料。Scatchard分析表明分子印迹复合材料基体中主要存在两类吸附位点,高亲和位点的平衡离解常数K和最大表观吸附量Qmax分别为0.5679 g/L和326.5 mg/g,而低亲和位点的K和Qmax分别为2.493 g/L和562.9 mg/g。分子印迹复合材料对吗啉的选择因子相对于甲基吗啉、乙基吗啉和甲基吗啉氧化物分别为2.47、2.48和2.24,均高于非印迹复合材料和MIL-101。将此材料作为吸附剂用于固相萃取时,其对模板的装载量高于非印迹复合材料。在优化条件下,采用分子印迹复合材料固相萃取水果粗提物中的吗啉时,单步洗脱中吗啉回收率达82.44%,具备较高选择吸附效能,而且这种复合材料可以多次重复使用。
猜你喜欢
中国食品学报(2022年8期)2022-09-07
江苏农业学报(2022年4期)2022-09-07
陶瓷研究(2022年3期)2022-08-19
广州化工(2022年3期)2022-02-24
中国果业信息(2021年5期)2021-12-05
云南画报(2021年10期)2021-11-24
世界农药(2020年12期)2021-01-04
农药科学与管理(2019年8期)2019-11-23
亚热带农业研究(2019年1期)2019-06-11
农药科学与管理(2019年12期)2019-05-20
