
先天性肝内胆管囊状扩张症合并常染色体隐性多囊肾的产前超声特征分析
2020-06-01 08:32:48王艺璇李天刚祁平安
临床超声医学杂志 2020年5期
马 斌 王艺璇 李天刚 冉 婕 鲁 琰 杨 磊 祁平安
随着产前超声仪器分辨力的提高及基因学诊断的不断开展,目前临床对胎儿肝脏及肾脏占位性病变的诊断率逐渐提高。先天性肝内胆管囊状扩张症又称Caroli病,其与常染色体隐性多囊肾(autosomal recessive polycystic kidney disease,ARPKD)均与位于染色体6p12的多囊肾/多囊肝病变1基因(PKHD1)突变相关[1]。超声不仅可以观察肝脏、肾脏声像图表现,而且可以实时动态监测病变进展情况,为临床早期诊断及治疗方式的选择提供可靠依据。本研究回顾性分析我院经基因检测结果确诊的5例Caroli病合并ARPKD胎儿的产前超声特征,以提高产前超声医师对该病的认识。
资料与方法
一、临床资料
选取2014年6月至2019年2月在我院诊断为Caroli病合并ARPKD的胎儿5例,孕22~29周,平均(25.0±3.2)周,孕妇年龄22~36岁,平均(28.0±2.4)岁。孕妇均无家族史,所有胎儿均在引产后经影像学或病理解剖证实。参照先天性胆总管囊肿Todani分型[2],Caroli病为TodaniⅤ型;参照美国妇产科超声诊断学标准[3],ARPKD为PotterⅠ型(婴儿型多囊肾),排除其他胆总管囊肿及肾脏囊性疾病类型。本研究经我院医学伦理委员会批准,所有孕妇均知情同意。
二、仪器与方法
使用GE Voluson E 10彩色多普勒超声诊断仪,RM6C凸阵探头,频率2~6 MHz,超声声强≤100 mW/cm2。孕妇取仰卧位或侧卧位,发现胎儿肝脏有囊状扩张、肾脏增大并弥漫性回声增强后由2名高年资超声医师进行肝脏及肾脏多切面扫查,测量胎儿肝脏从横膈右侧穹窿部到肝脏下缘的长度、肾脏大小、肝内胆管扩张宽度及羊水指数,并对胎儿其他系统进行详细检查,观察是否合并其他系统畸形。
对诊断为Caroli病合并ARPKD的胎儿,建议孕妇进行产前诊断咨询,引产胎儿追踪其解剖及其他影像学结果。若家属想进一步明确诊断,则对引产胎儿取部分脐带组织,应用第二代测序方法对囊性肾脏病PKHD、PKD基因进行全外显子测序,并取其父母的外周血标本,进行Sanger法测序验证。分析记录肝脏及肾脏超声病变胎儿的胎龄、性别、超声表现、合并异常及临床处理结局。
结 果
5例孕妇均选择引产终止妊娠并行基因学检测,均检出PKHD1基因存在缺陷。5例Caroli病合并ARPKD胎儿的临床资料、产前超声和基因学诊断结果及临床处理结果见表1;超声表现、引产后大体尸检表现见图1~4。
5例胎儿产前超声均清晰显示为肝脏内不同程度肝内胆管扩张,双肾体积弥漫性增大、回声增强,皮髓质界限不清。合并羊水过少3例,单脐动脉1例,室间隔缺损1例,单侧下肢长骨发育不良并足内翻1例。

表1 5例Caroli病合并ARPKD胎儿的临床资料、产前超声和基因学诊断结果及临床处理结果

图1 Caroli病合并ARPKD胎儿超声图(箭头示肝内胆管囊状扩张)

图2 引产后大体标本图(箭头示肝内胆管囊状扩张内部可见门静脉分支)
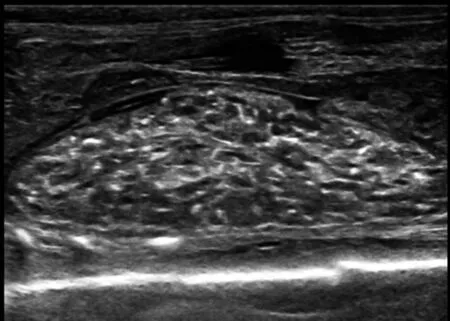
图3引产后大体标本超声检查显示肾实质内弥漫分布的小囊状无回声区
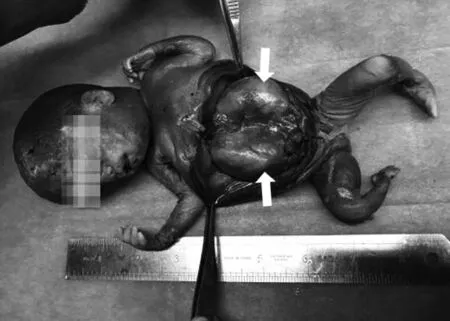
图4 引产后大体标本图(箭头示双肾体积明显增大)
讨 论
Caroli病于1958年法国医生Caroli首先描述而得名,常根据是否合并肝纤维化分为Ⅰ型(单纯型)和Ⅱ型(硬化型),后者则称为Caroli综合征[4]。ARPKD的肾脏病变为肾髓质集合管囊性变,重者皮髓质均受累,最终导致高血压病和慢性肾功能不全。Caroli病和ARPKD常同时发生,并发率高达71%[5-6]。两者均是由于PKHD1基因突变导致的早发性肝肾囊性纤维化疾病[7],表现为胆管板发育障碍出现胆管板畸形,并影响肾小管上皮细胞的分化、增殖和凋亡,从而出现胆管和肾小管的囊性病变,组织病理学特征为胆管上皮增生和囊状扩张。
Caroli病合并ARPKD临床表现隐匿,多见于儿童和青年,80%患者在30岁以前发病,婴儿期常以肝、肾肿大为唯一临床表现。单纯Caroli病胎儿期发病罕见。单纯ARPKD胎儿期发病较为常见,其肾脏改变引起肾脏功能减低,导致羊水产生过少,常伴发肺发育不良等其他结构畸形,围生期死亡率约30%~40%[8]。ARPKD的产前超声图像特征性明显,但肝内超声病变常难以发现,原因为胎儿期肝内细微结构声像图显示欠清晰,不能及时发现肝内早期扩张胆管,同时对肝脏增大测量较为困难。临床研究[6,8]认为,Caroli病合并ARPKD胎儿的肾功能减低明显早于肝功能减低。本研究5例胎儿均出现肾脏增大、回声增强等超声征象,笔者认为若在产前超声检查时已经发现明显的肝内胆管囊状扩张,说明疾病进入进展期。
Caroli病产前诊断主要依靠超声等影像学检查,其典型的声像图表现为肝内可见散在或串珠样分布的囊性回声,内可见点状或带状门静脉或肝动脉的高回声,即“中心点征”,若囊肿较大,则需与肝内其他囊性病相鉴别。ARPKD的典型产前超声表现为双肾体积明显增大,可充满全腹,较正常肾体积大12~16倍[9],实质内可见细小密集的囊性回声,因大量囊性结构造成的界面反射,实质回声弥漫性增强,中央集合系统仅见痕迹,膀胱常不显示伴羊水过少,严重者还合并其他部位畸形如肺部发育不全、脸部多发性畸形、骨骼畸形等。本研究5例Caroli病合并ARPKD胎儿的肝、肾双器官病变的超声表现典型,合并其他异常主要为羊水过少、单脐动脉、室间隔缺损及单侧下肢长骨发育不良并足内翻,分析因胎儿肾脏功能减低,导致中孕期或晚孕期出现羊水过少,出生后由于肺发育不全难以存活,故本研究5例孕妇均选择引产。
Caroli病合并ARPKD为常染色体隐性遗传,目前PKHD1基因已报道突变300余种,主要为错义突变,少数导致蛋白截断突变[10]。国外研究[11-12]发现该病基因型及临床表现之间缺少相关性,部分患者基因诊断明确时仍未出现临床症状。但本研究5例产前超声考虑Caroli病合并ARPKD胎儿,基因检测均发现PKHD1基因突变,说明本病基因型与临床表现之间关系的复杂性,推测可能有其他基因参与了PKHD1基因的表达调控,同时表明通过典型的特征性声像图表现,可做出准确的初步诊断。因此欧美专家提出的常染色体隐性多囊肾病的诊断和治疗专家建议[13]指出,现阶段该病的诊断主要依靠影像学检查,基因诊断作为重要补充方法,可以对复杂影像学表现胎儿进行明确及鉴别诊断。
综上所述,超声医师可以依据胎儿肝脏及肾脏的特征性声像图特征,做出Caroli病合并ARPKD的初步诊断。超声作为早期诊断该病的有效手段,当发现其中一种病变时,应注意观察其余一种病变,避免漏误诊,同时需要结合羊水、染色体异常等多方面因素,为临床及早干预及选择治疗方式提供可靠依据。
猜你喜欢
江苏卫生保健(2021年9期)2021-03-27 16:25:04
家庭医药(2021年2期)2021-03-09 06:48:09
保健医苑(2020年6期)2020-12-04 01:33:11
中国临床医学影像杂志(2019年1期)2019-04-25 06:49:38
西藏科技(2016年8期)2016-09-26 09:00:48
海南医学(2016年8期)2016-06-08 05:43:00
中国卫生标准管理(2015年7期)2016-01-15 03:58:37
科学之友(2014年20期)2014-12-23 18:55:56
长江大学学报(自科版)(2014年12期)2014-03-20 13:21:41
中国全科医学(2014年14期)2014-01-28 09:06:02
