
压水堆二回路工况下碱化剂对结构材料腐蚀的影响
2020-05-30 01:36:18曹林园杨明馨皮立新
原子能科学技术 2020年5期
曹林园,王 辉,杨明馨,皮立新,张 鹏
(中国原子能科学研究院 反应堆工程技术研究部,北京 102413)
压水堆核电站二回路系统有效的水化学控制能降低设备材料的腐蚀[1]。一般通过在除盐水中加入联氨等除氧剂除氧(溶解氧≤10 μg/L),加入氨(NH3·H2O)等碱化剂调节pH值约为9.6~9.8(25 ℃)。但由于NH3·H2O的气液分配系数大而使液相pH值低,从而导致各疏水系统及所有蒸汽管道出现严重流动加速腐蚀现象。近年来有研究发现,乙醇胺(ETA)、二甲胺(DMA)等有机胺具有碱性强、分子量小、热稳定性好、气液分配系数适当的特点,可作为NH3·H2O的替代品用于二回路水化学控制[2];进一步的研究表明,与单独使用ETA相比,复合碱化剂可进一步降低二回路中Fe的含量。复合型碱化剂既能充分发挥每种碱化剂的优势,又能避开单一胺的不足,以致在应用中能较好地调控二回路水质,又能与二回路设备实现良好的兼容[3-5]。目前,美国约有75%的压水堆核电站采用ETA或将ETA与其他有机胺复合使用作为二回路的pH值调节剂[6-10]。而国内现役核电站大多采用添加NH3·H2O的方法,仅秦山一期采用添加ETA的控制方法,秦山二期采用NH3·H2O+ETA的混合控制模式[11]。从运行经验看,复合碱化剂能使给水中Fe含量下降30%,气水分离再热器疏水Fe含量下降80%以上。
当前在我国正倡导大力发展核电、重金引进AP1000与自主研发CAP1400的形势下,更需要系统研究和评估先进水化学工况对蒸汽发生器气液系统材质的相容性和对新工艺的适应性。复合碱化剂水化学工况在国外得到成功应用[12],但国内在役压水堆核电机组使用较少,说明复合碱化剂水化学工况的控制要点、方法,甚至作用机理仍未掌握。本文以低合金钢A508Ⅲ和A106Gr.B为研究对象,考察在ETA+DMA、ETA及NH3·H2O等不同碱化剂条件下材料的腐蚀性能与机理,为二回路碱化剂技术优化提供必要的数据支持。
1 试验
1.1 金属材料
实验材料为国产A508Ⅲ低合金钢和A106Gr.B碳钢,其成分列于表1。将材料线切割成20 mm×15 mm×2 mm的片状样品,用SiC砂纸依次打磨至镜面光亮,用无水乙醇超声清洗、干燥。
1.2 仪器
扫描电镜/能谱仪(SEM/EDS):扫描电镜型号JSM-6400,能谱仪为美国EDAX公司产品。纳米扫描俄歇系统,型号PHI-700,日本ULVAC-PHI公司。同轴电子枪和CMA能量分析器,电子枪高压为5 kV,能量分辨率为0.1%;入射角为30°,分析室真空度优于5.2×10-7Pa,扫描型Ar+枪,标样为热氧化SiO2/Si;溅射速率为66 nm/min。
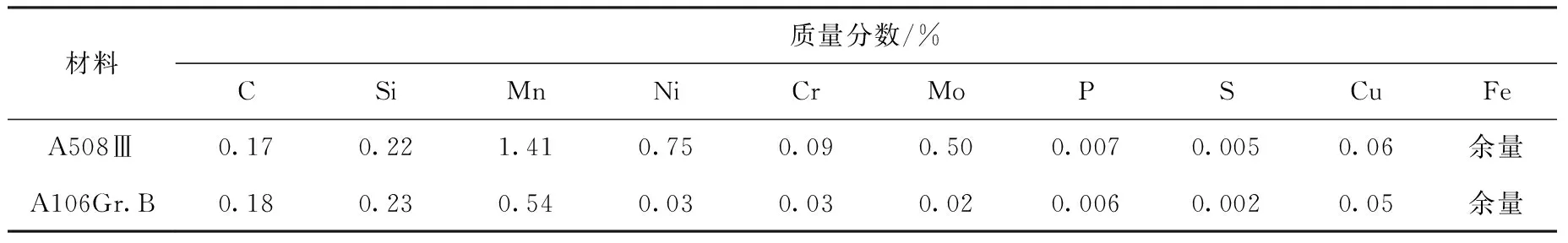
表1 试验用金属材料的主要成分及其含量Table 1 Main component and content of metal material for testing sample
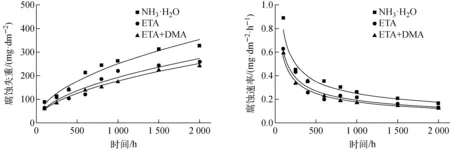
图1 A106Gr.B钢的腐蚀失重量和腐蚀速率随时间的变化Fig.1 Corrosion weight loss and corrosion rate of A106Gr.B vs. time
1.3 试验方法
静态浸泡试验依据GB 10124—1988《金属材料实验室均匀腐蚀全浸试验方法》进行。相容性试验采用高温高压釜,试验釜容积为5 L,设计温度为350 ℃,设计压力为15.5 MPa。控温精度为±1 ℃。试验条件为相同pH值(9.8)的二回路水化学条件,分别为含ETA(20 mg/L)溶液、5 mg/L ETA+0.5 mg/L DMA溶液和5 mg/L NH3·H2O,溶解氧浓度控制在10 μg/L以下(添加联氨除氧),溶液均由去离子水及分析纯试剂配制而成。腐蚀试验的试验温度为(280±1) ℃,从室温升温至105 ℃进行热力除氧,然后向釜内注入一定量的ETA和联氨,逐渐升温至目标温度,升温过程约4 h。实验介质7 d更换1次。试验的面容比为20 mL/cm2。每个时间点取2个平行试样。
腐蚀速率分析及失重分析过程如下:1) 取出腐蚀后的试样,干燥24 h,测量质量;2) 对试样进行脱膜处理,测量腐蚀后试样脱膜质量;3) 采用称重的方式,利用腐蚀前与腐蚀试样脱膜后的质量差随时间的变化来对比不同碱化剂对材料腐蚀失重及腐蚀速率的影响。脱膜方法详见文献[13]。
2 结果分析与讨论
图1为A106Gr.B试样在NH3·H2O、ETA和ETA+DMA环境下的腐蚀失重情况。可看出,3种环境下试样均发生了失重,且随着时间的延长,失重量增大;经历2 000 h腐蚀试验后,失重量分别为327、259、242 mg/dm2,说明试样在NH3·H2O环境下的腐蚀更严重。在同一时间下,试样在NH3·H2O环境下的腐蚀失重量均最大,主要是由于NH3·H2O的挥发性较大,试验温度下大部分NH3·H2O挥发进入气相,导致液相NH3·H2O浓度下降,引发试样腐蚀加剧。对比ETA和复合胺环境下的试样可知,试验初期的600 h内,腐蚀失重量随腐蚀时间呈现先增加后降低的趋势,试验后期复合胺环境下试样的腐蚀失重量低于ETA环境的。根据金属腐蚀的动力学规律,碳钢及低合金钢在不同碱化剂的二回路环境中的腐蚀失重量y与腐蚀时间x满足y=ax1/2关系[14]。对A106Gr.B钢的腐蚀失重数据进行拟合,得到以下关系式:NH3·H2O,y=7.913 83x1/2;ETA,y=6.099 98x1/2;ETA+DMA,y=5.617 96x1/2。历经2 000 h腐蚀后,试样在3种环境下的腐蚀速率分别为0.163 5、0.129 5、0.121 0 mg/(dm2·h),ETA与ETA+DMA中的腐蚀速率相对于NH3·H2O分别下降22.92%和29.01%。
考察了A508Ⅲ钢与复合碱化剂的相容性。不同水化学环境下其均匀腐蚀失重量和腐蚀速率与腐蚀时间的关系示于图2。3种环境下,A508Ⅲ钢的腐蚀失重量随时间延长而逐渐增大。对其腐蚀失重量y与时间x的关系进行拟合,所得数学表达式如下:NH3·H2O,y=7.488 82x1/2;ETA,y=5.469 86x1/2;ETA+DMA,y=3.539 24x1/2。还可看出,在腐蚀初期的100 h内,腐蚀速率较大。其中ETA环境下的腐蚀速率高达0.76 mg/(dm2·h)。随着腐蚀时间的延长,腐蚀速率逐渐降低。3种环境下的腐蚀速率大小为:NH3·H2O>ETA>ETA+DMA。腐蚀时间达2 000 h时,3种环境下的腐蚀速率分别为0.15、0.11、0.087 mg/(dm2·h)。经计算,A508Ⅲ在ETA和ETA+DMA环境下的腐蚀速率相对于NH3·H2O下分别下降26.96%和52.74%。由此可见,复合碱化剂能有效降低A508Ⅲ材料的腐蚀速率。
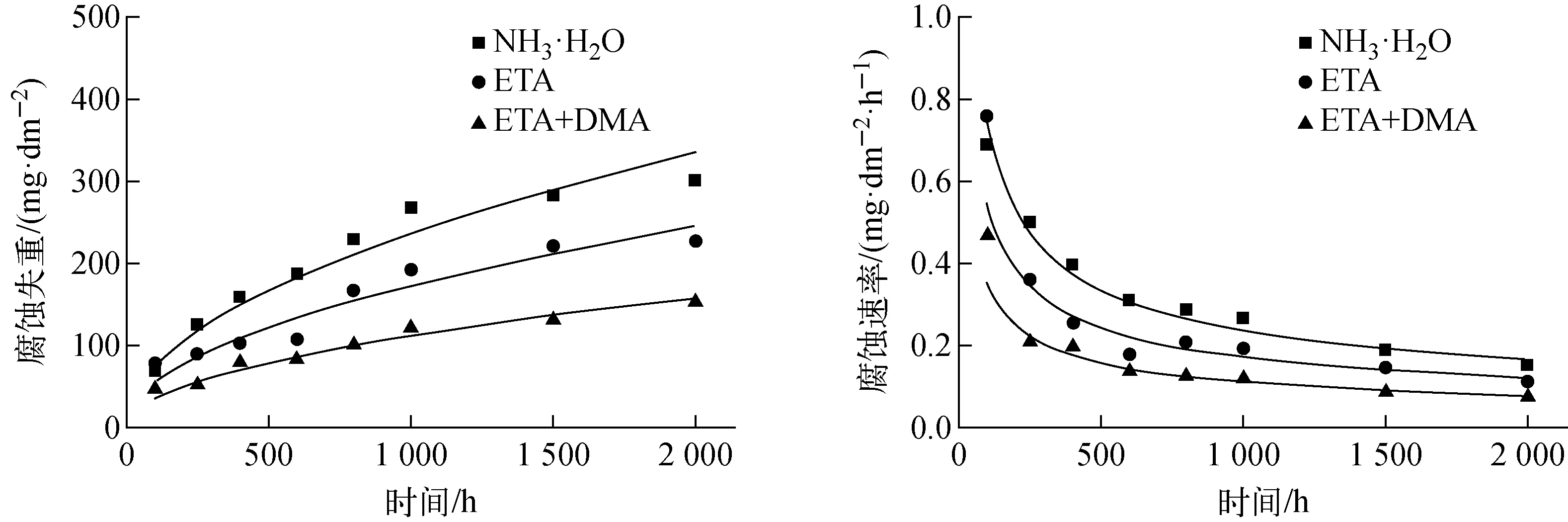
图2 A508Ⅲ钢的腐蚀失重量和腐蚀速率随时间的变化Fig.2 Corrosion weight loss and corrosion rate of A508Ⅲ steel vs. time
为进一步考察碱化剂对试样氧化膜结构的影响,利用扫描电镜对其进行了微观分析。图3为A106Gr.B钢在NH3·H2O、ETA和ETA+DMA环境下腐蚀1 000 h后的SEM图像。可见,3种环境下试样表面都生成了明显的氧化膜。在放大100倍下,NH3·H2O环境下的氧化膜表面呈现较多大颗粒块状氧化物,氧化物中间有些许空隙,分布也不均匀(图3a);至放大1 000倍后,发现这些较大的颗粒是由许多呈八面体的细小颗粒堆积而成,这些细小颗粒的尺寸在200~500 nm之间,结构较为疏松(图3b)。与NH3·H2O环境相比,ETA环境下形成的氧化膜结构更加致密、均匀,氧化物颗粒尺寸更均一(图3c);在放大1 000倍条件下观察,氧化物膜结构孔隙较多,结构疏松(图3d)。由图3e可知,在放大100倍情况下,ETA+DMA环境下氧化膜表面是1层分散均匀的颗粒状物质,结构较为疏松;进一步在放大1 000倍下观察(图3f),发现这些疏松的氧化物颗粒尺寸可达μm级,零散分布在氧化膜表面,在这些大颗粒下面是1层结构致密的小粒径氧化物颗粒组成的膜结构,这可能是阻止试样进一步腐蚀的主要原因。

a,b——NH3·H2O;c,d——ETA;e,f——ETA+DMA图3 A106Gr.B试样在3种碱化剂中腐蚀1 000 h后的SEM图像Fig.3 SEM image of oxide film of A106Gr.B specimen immersed in different alkalizers for 1 000 h
图4为A508Ⅲ钢在3种环境下腐蚀1 000 h后的形貌。由图4可知,3种条件下试样表面均可观察到清晰的打磨痕迹,在其表面生成的氧化膜由分布均匀的细小颗粒组成。而在ETA和ETA+DMA条件下有较大的氧化物颗粒。进一步放大图像可看出,NH3·H2O环境下的氧化膜较为粗糙,氧化膜颗粒之间有较大的孔洞,冷却剂通过这些孔洞进入氧化膜内部,与基体材料发生固相反应,加快腐蚀速率。ETA+DMA条件下的氧化膜相对较为均匀。

a,b——NH3·H2O;c,d——ETA;e,f——ETA+DMA图4 A508Ⅲ钢在3种环境下腐蚀1 000 h后的SEM图像Fig.4 SEM image of oxide film of A508Ⅲ specimen immersed in different alkalizes for 1 000 h
大颗粒氧化物下面是结构紧密的氧化膜,经Nano Measurer1.2软件测得NH3·H2O环境下,表层大颗粒氧化物尺寸在200~300 nm之间,ETA环境下的氧化物颗粒较NH3·H2O下的略大,出现了大片状的氧化物颗粒,ETA+DMA条件下尺寸进一步增大,在1~3 μm之间。说明试样表面氧化膜的生成与其水化学环境中的胺分子有关。从NH3·H2O到复合胺,相同pH值下水化学环境中的胺分子逐渐减少,导致吸附于氧化物颗粒表面的胺分子数目减少,可能会影响氧化物颗粒的生长模式。
为进一步分析复合碱化剂对氧化膜结构的影响,以A508Ⅲ为例,用XPS和AES技术对其氧化膜的成分进行深入分析与比较。XPS结果显示:试样氧化膜的主要组分是Fe和O,有微量N元素。进一步对Fe元素分峰,结果如图5所示。NH3·H2O环境下,氧化膜中Fe2+含量远高于Fe3+含量,通过积分得到二者含量分别约为90%和10%,推测其氧化物中含有大量Fe(OH)2。而ETA+DMA条件下,两种Fe离子的含量经拟合计算分别为59%和41%,可推测试样在该条件下形成的氧化膜内含有大量的Fe3O4,利于形成致密结构的氧化膜[15-16]。
对A106Gr.B试样在NH3·H2O和复合碱化剂环境中形成的氧化膜厚度进行AES表征,结果示于图6。随溅射深度的增加,Fe、N元素含量逐渐下降。溅射初期,两种环境中的试样氧化膜成分主要有Fe、O及少量的C和N,Fe/O比基本为4/3,说明氧化膜内主要是Fe的氧化物和吸附的少量胺分子。当溅射深度达1.7 μm后,复合碱化剂环境下试样的氧化膜中Fe含量开始升高,O含量逐渐下降,直到3 μm后Fe含量达92%以上,O、C和N总量不足5%,说明到达试样基底。而NH3·H2O环境下的氧化膜溅射深度约为3 μm时,才开始出现O含量下降和Fe含量升高的趋势,说明NH3·H2O环境下的氧化膜厚度大于复合碱化剂环境下的。
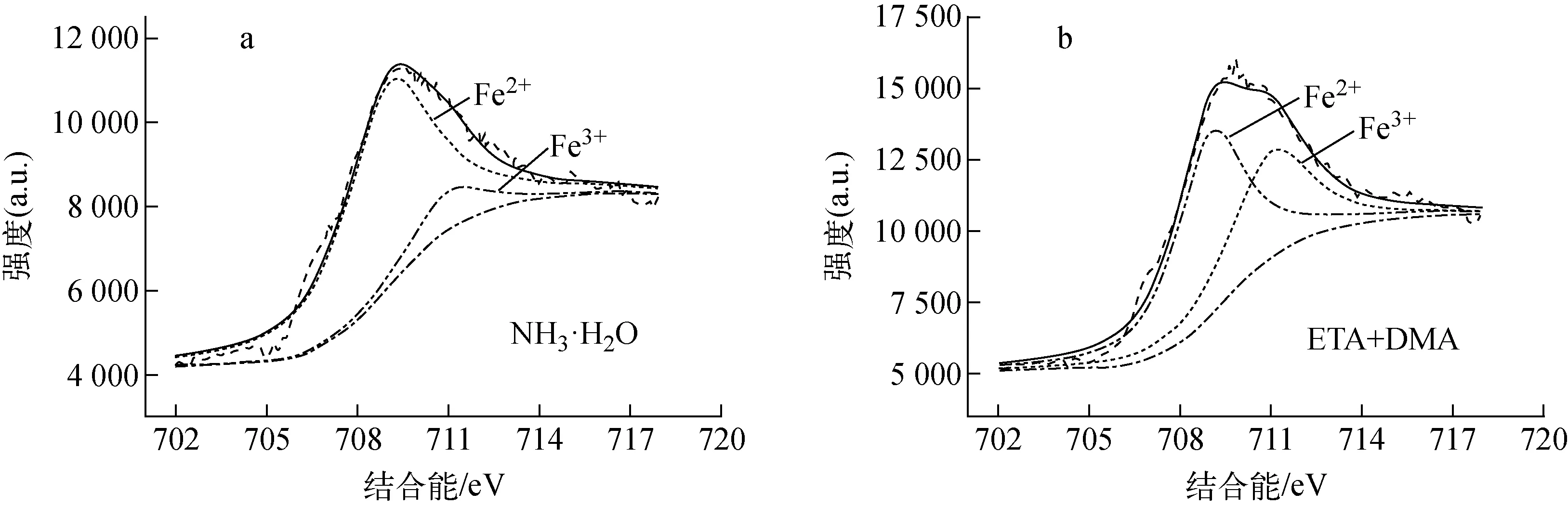
图5 A508Ⅲ在碱化剂中腐蚀2 000 h后氧化膜XPS中Fe元素的分谱拟合曲线Fig.5 Fe 2p spectra of oxide film formed on A508Ⅲ in NH3·H2O and ETA+DMA for 2 000 h
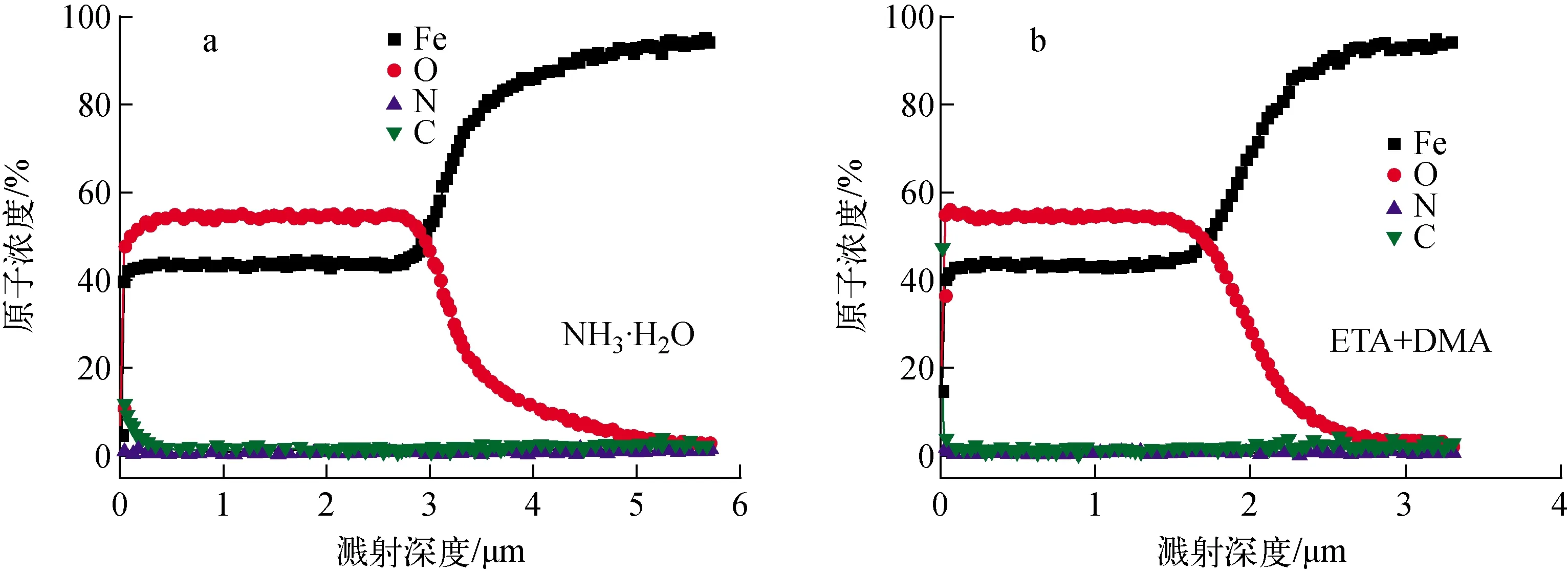
图6 A106Gr.B试样在NH3·H2O和ETA+DMA中腐蚀2 000 h后氧化膜的AES谱Fig.6 AES spectrum of oxide film of A106Gr.B specimen immersed in NH3·H2O and ETA+DMA for 2 000 h
280 ℃下A106Gr.B和A508Ⅲ试样在NH3·H2O、ETA和ETA+DMA中的极化曲线示于图7。从图7a可看出,A106Gr.B试样在3种环境下的极化行为很相似,在整个阳极极化区,电流密度I随电位E的增加逐渐增大,未观察到明显的钝化区,说明碱化剂的改变对试样的极化行为并无太大影响[17]。当电位大于0 V后,电流密度逐渐达到相似数值。经拟合计算,试样在NH3·H2O、ETA和ETA+DMA中的自腐蚀电位十分接近,依次为-0.755、-0.718、-0.70 V。可见,ETA和ETA+DMA条件下的腐蚀电位相当,较在NH3·H2O环境中升高约35 mV,说明试样在ETA或复合碱化剂环境中的耐蚀性略高。其相应的自腐蚀电流密度分别为3.39、2.95、2.81 μA/cm2。从图7b可看出,A508Ⅲ试样在3种环境下的极化曲线变化趋势与A106Gr.B试样的极为类似。经计算拟合求得试样在3种环境下的自腐蚀电位分别为-0.77、-0.68、-0.67 V。相应的自腐蚀电流密度分别为7.41、4.08、3.63 μA/cm2。在整个极化过程中,试样在ETA和ETA+DMA下的腐蚀电流密度均小于NH3·H2O环境,尤其是当电位从-0.5 V升至0.5 V区间,腐蚀电流密度随电位升高的增幅变得十分缓慢,与NH3·H2O环境下相比,腐蚀电流密度降低约1个数量级,说明材料的腐蚀速率下降。由此可见,复合碱化剂下试样的腐蚀电流密度最小,腐蚀趋势最轻。这与失重腐蚀试验结果一致。
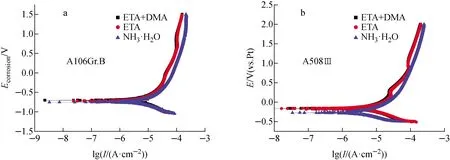
图7 A106Gr.B和A508Ⅲ在3种环境中的极化曲线Fig.7 Linear polarization curve of A106Gr.B and A508Ⅲ specimen immersed in different alkalizers

另一方面,低合金钢在高温高压下的腐蚀主要是Fe被氧化生成Fe3O4的过程。根据点缺陷模型[20],腐蚀产物Fe3O4是氧化膜与溶液界面处和金属与氧化膜界面处的化学反应形成的。在腐蚀过程中金属离子空位在基体与氧化膜处积累会造成氧化膜和金属基体的脱离[21]。就Fe在一系列pH值的高温溶液中而言,pH值越高,氧化膜与基体处金属离子空位的浓度越低,越不容易形成空位累计而致使腐蚀产物脱落。ETA和DMA在高温高压下的相对挥发性均低于NH3·H2O,因此复合碱化剂环境下高温液相中胺的浓度较NH3·H2O环境下的高。而室温下pH值均为9.8的NH3·H2O、ETA和ETA+DMA溶液在280 ℃时的理论pH值分别为6.28、6.45和6.50,显然复合碱化剂的碱性最强。在这种环境下,金属离子在溶液中的溶解度将降低,金属离子空位源减少,在一定程度上抑制了金属的腐蚀,进一步说明复合碱化剂环境下材料的耐蚀性最好。
3 结论
通过高温高压腐蚀试验考察了压水堆二回路工况下A106Gr.B和A508Ⅲ钢分别在ETA+DMA、ETA和NH3·H2O碱化剂中的均匀腐蚀和电化学腐蚀性能,利用SEM、XPS和AES等技术研究了氧化膜的结构,分析了复合碱化剂下材料耐腐蚀性能提高的原因,得出以下主要结论。
1) 经过2 000 h腐蚀试验后,A508Ⅲ试样在NH3·H2O、ETA和ETA+DMA中的均匀腐蚀速率分别为0.15、0.11、0.087 mg/(dm2·h),ETA+DMA环境下较NH3·H2O环境下降低约42%。对于A106Gr.B材料,ETA+DMA环境下的腐蚀速率相对于NH3·H2O下降约29.01%。
2) 试样在碱化剂中形成的氧化膜基本都是Fe3O4,但复合碱化剂环境下的氧化膜表面颗粒物相对较均匀平整,结构更致密,厚度明显减薄。
3) 复合碱化剂中的胺分子易吸附于氧化膜表面,降低了金属氧化反应的活化能;DMA和ETA的相对挥发性小于NH3·H2O,液相冷却剂pH值提高,氧化反应减缓。
4) 复合碱化剂调控二回路冷却剂pH值可提高低合金钢材料的耐蚀性。
猜你喜欢
北方交通(2022年8期)2022-08-15 09:47:48
黑龙江水利科技(2020年8期)2021-01-21 09:26:50
中国油脂(2020年11期)2020-11-13 09:41:14
祝您健康·养生堂(2020年1期)2020-04-19 10:04:48
现代畜牧科技(2019年11期)2019-01-06 02:15:01
河北地质(2016年1期)2016-03-20 13:51:57
中国煤层气(2015年5期)2015-08-22 03:26:32
建筑材料学报(2015年3期)2015-02-28 02:36:31
当代畜禽养殖业(2014年2期)2014-08-22 02:38:44
中国科技博览(2014年28期)2014-08-06 14:21:05
