
PI3K抑制剂联合用药抗肿瘤的研究进展
2020-05-28 10:47:32吕鑫钰张亚梅孔德新
食品与药品 2020年2期
吕鑫钰,张亚梅,刘 璐,孔德新
(1.天津天狮学院 医学院,天津 301700;2.天津医科大学 药学院,天津 300070)
目前肿瘤的临床治疗依然以手术、化疗药物为主,但进入二十一世纪以来分子靶向药物治疗正扮演着越来越重要的角色。分子靶向药物相较于传统的细胞毒性药物,因为以肿瘤细胞不同于正常细胞的特征为靶点,由盲目攻击变为有的放矢,在发挥强大的抗肿瘤作用同时,减少了对正常器官和组织的毒副作用,从而改善患者生存质量,因此成为肿瘤治疗药物研发的热点。
磷脂酰肌醇3-激酶(PI3K)是PI3K/AKT/mTOR信号通路的起始节点,在肿瘤细胞的增殖、迁移、侵袭、血管生成等过程中起重要作用,成为肿瘤靶向治疗的热门靶点。本文介绍了几种具有代表性的PI3K抑制剂单独和联合用药的抗肿瘤临床和临床前研究进展。
1 PI3K抑制剂与肿瘤
PI3K由调节亚基和催化亚基构成,根据其结构特点可分为I类、II类、III类,其中I类PI3K因研究较多,为通常人们所提到的PI3K。这类PI3K是一种异二聚激酶,根据调节亚基和上游激活因子的不同又可分为IA类和IB类。IA类包含PI3Kα、PI3Kβ、PI3Kδ 3种亚型。而IB类为PI3Kγ。其中PI3Kα和PI3Kβ在机体内广泛表达,而PI3Kδ和PI3Kγ主要表达于造血细胞。在肿瘤的发生过程中,PI3Kα变异发挥重要作用。除此之外,PI3Kα与葡萄糖代谢、PI3Kβ与血小板激活、PI3Kδ和PI3Kγ与免疫系统疾病密切相关[1]。I类PI3K在未被激活状态下,其调节亚基p85和催化亚基p110产生相互作用,导致p110激酶活性被抑制。当被受体酪氨酸激酶(receptor tyrosine kinase,RTK)或G蛋白偶联受体(G-protein coupled receptor,GPCR)激活后,p110激酶活性的抑制被解除,从而催化磷脂酰肌醇4,5-二磷酸(PIP2)磷酸化为磷脂酰肌醇3, 4, 5-三磷酸(PIP3),而这一过程可被PTEN逆转。PIP3作为第二信使,可募集AKT和 PDK1等蛋白激酶至细胞膜并激活,此外AKT还可被PDK1和mTORC2激活,继而激活下游Akt/mTOR通路产生促进细胞周期进展、维持细胞存活、调控细胞生长、促进血管生成等作用(图1)。而PTEN作为典型的抑癌基因,其缺失是导致PI3K过度激活的常见原因[2]。

图1 PI3K与肿瘤的发生发展和转移密切相关
2 PI3K抑制剂单独用药抗肿瘤
目前PI3K抑制剂分为3类,第一类为PI3K/mTOR双重抑制剂如NVP-BEZ235,第二类为泛PI3K抑制剂如ZSTK474,第三类为PI3K亚型特异性抑制剂如Alpelisib(阿培利司)。迄今为止已有4个PI3K抑制剂被批准上市(表1),包括吉利德制药研发的Idelalisib(艾德拉利司),拜耳制药研发的Copanlisib(库潘利司),武田研发的Duvelisib(杜沃利司)以及诺华制药研发的Alpelisib。
2.1 NVP-BEZ235
NVP-BEZ235是一种PI3K/mTOR双重抑制剂,由诺华制药研发,是第一个进入临床试验的PI3K抑制剂,目前NVP-BEZ235针对晚期实体瘤、胰腺神经内分泌肿瘤的抗肿瘤研究已在美国进入临床试验阶段。另外,Deng等[3]研究证实了NVP-BEZ235针对急性髓系白血病的抗肿瘤效果,显示NVPBEZ235通过上调抑癌基因miR-1-3p表达,抑制了急性髓系白血病两种多药耐药细胞系HL-60/VCR和K562/ADR的增殖和迁移,并提高了化疗药物的敏感性。NVP-BEZ235在抗肿瘤的机制中也涉及细胞自噬过程,Ma[4]等报道NVP-BEZ235抑制多发性骨髓瘤细胞的增殖和促进肿瘤细胞凋亡,并可诱导肿瘤细胞自噬过程,主要涉及mTOR2-Akt-FOXO3a-BNIP3通路。
2.2 Idelalisib
Idelalisib(Zyd eligTM)是一种口服PI3Kδ抑制剂,2014年7月由美国食品药品监督管理局(FDA)批准上市,与利妥昔单抗联合用药,用于治疗复发/难治性慢性淋巴细胞白血病、滤泡性淋巴瘤和小淋巴细胞淋巴瘤,为第一个批准上市的PI3K抑制剂。此外,Idelalisib 针对非霍奇金淋巴瘤、慢性淋巴细胞白血病等的抗肿瘤研究已在日本、美国进入临床试验阶段[5]。Yang[6]等报道Idelalisib 可诱导结肠癌细胞凋亡,且该作用可能与对Bcl-2家族成员PUMA的影响有关,因此作者提出PUMA可作为一种生物标志物预测Idelalisib的抗结肠癌疗效。

表1 已被批准上市的PI3K抑制剂
2.3 Copanlisib
Copanlisib(AliqopaTM)是一种强效泛PI3K抑制剂,于2017年5月被FDA批准上市,主要用于治疗成年患者(该患者至少已接受过两次系统治疗)的复发性难治性滤泡性淋巴瘤[7]。此外,Copanlisib静脉注射用于惰性和侵袭性淋巴瘤的治疗也已经进入II期临床试验,且表现出良好的临床疗效以及可控的毒性[8]。针对晚期肿瘤和非霍奇金淋巴瘤的研究已完成I期临床试验。
2.4 Duvelisib
Duvelisib(CopiktraTM)于2018年9月24日在美国首次获得批准,用于治疗患有复发性或难治性慢性淋巴细胞白血病及小细胞淋巴瘤的成年患者。该药是由武田制药旗下的Intellikine公司研发的,是一种口服小分子选择性PI3Kγ和PI3Kδ双重抑制剂。目前Duvelisib用于治疗复发或难治性滤泡性淋巴瘤(至少进行过两次全身治疗的成年患者)获得加速批准[9]。Duvelisib用于复发/难治性淋巴瘤的I期临床试验已在日本完结。在以往的I期临床试验中对给予非霍金淋巴瘤患者(对利妥昔单抗和化疗或放射免疫治疗无效)口服Duvelisib进行了安全性和有效性评估,近期开展的一项II期临床试验再次证实了口服Duvelisib的临床效果和可控的安全性,与I期临床试验结果保持一致,提示口服Duvelisib在治疗难治性非霍奇金淋巴瘤的可能性[10]。关于Duvelisib对实体瘤的抗癌效果已有一些临床前研究。
2.5 Alpelisib
Alpelisib (PiqrayTM)(BYL719)是特异性PI3Kα抑制剂,于2019年5月24日由FDA批准,与氟维司群联合用药用于治疗绝经后女性或男性,伴有激素受体(HR)阳性(HR+),人表皮生长因子受体2(HER2)阴性(HER2-),并伴有PI3KCA突变的晚期、转移性乳腺癌患者。同时由QIAGEN公司研发的血浆PIK3CA突变检测试剂盒已被FDA批准作为接受Alpelisib治疗患者的辅助诊断[11]。Alpelisib用于晚期实体瘤的治疗已在日本进入I期临床试验。此外,有研究结果表明Alpelisib可抑制骨肉瘤细胞的增殖、迁移、肿瘤血管生成和骨细胞分化而达到抗肿瘤效果[12]。
2.6 ZSTK474
ZSTK474是Zenyaku研发的一种口服的泛PI3K抑制剂,其针对晚期实体瘤的临床试验已分别在美国和日本进行。在体外试验中,ZSTK474可抑制多达39种人源癌细胞的生长。我们的研究表明,ZSTK474有抑制肿瘤血管生成的作用,其机制包括对肿瘤血管内皮细胞生长和迁移的直接抑制和抑制VEGF(血管内皮生长因子)分泌作用。对于不同的人癌细胞ZSTK474均表现出显著的细胞周期阻滞作用,但未表现出明显的诱导肿瘤细胞凋亡作用[13]。另外,ZSTK474可抑制前列腺癌细胞的迁移和侵袭,并能抑制前列腺癌骨转移[14-15]。PI3K/AKT/mTOR通路在细胞自噬的调控中扮演着重要的角色。我们的研究表明ZSTK474对于乳腺癌细胞MCF-7具有明显的诱导细胞自噬作用[16]。
3 PI3K抑制剂联合用药抗肿瘤
细胞信号转导通路本质上是细胞分子水平上的功能调节,是机体细胞内外生理功能调节的重要基础环节,复杂庞大,且各个信号通路之间相互影响而形成信号网络系统,涉及基因表达、蛋白质相互作用等重要过程。PI3K为PI3K/AKT/mTOR信号通路的重要节点分子,抑制PI3K产生抗肿瘤活性的同时,也会通过信号通路反馈带来代偿性激活,从而引起耐药性及副作用的产生。目前已有几十种PI3K抑制剂进入临床试验或已被批准上市,但单独用药表现出的疗效不如预期好,主要体现在抗肿瘤作用局限、副作用、耐药性等方面。而联合用药一方面可提高疗效,另一方面可通过降低两种药物的剂量从而减轻副作用,因此PI3K抑制剂与化疗药物、其他分子靶向药物、以及放疗的联合治疗近年来成为热点,目的在于寻找更有效且安全的抗肿瘤用药组合。
3.1 NVP-BEZ235联合用药
顺铂为常用化疗药物,但顺铂的细胞毒副作用及耐药性的产生限制了其临床应用。近期研究结果表明,在体内和体外试验中,NVP-BEZ235均可通过诱导肿瘤细胞自噬提高肿瘤细胞对顺铂的敏感性,产生协同的抗肿瘤活性[17]。另外两项研究证实该联合用药方案对于非小细胞肺癌和下咽鳞癌产生协同的抗肿瘤活性[18-19]。此外NVP-BEZ235与醋酸阿比特龙联合用药用于转移性去势耐药前列腺癌的治疗已进入Ⅰ期临床试验,但结果表明患者耐受性较差,出现了不可接受的药物毒性,试验被迫终止[20]。
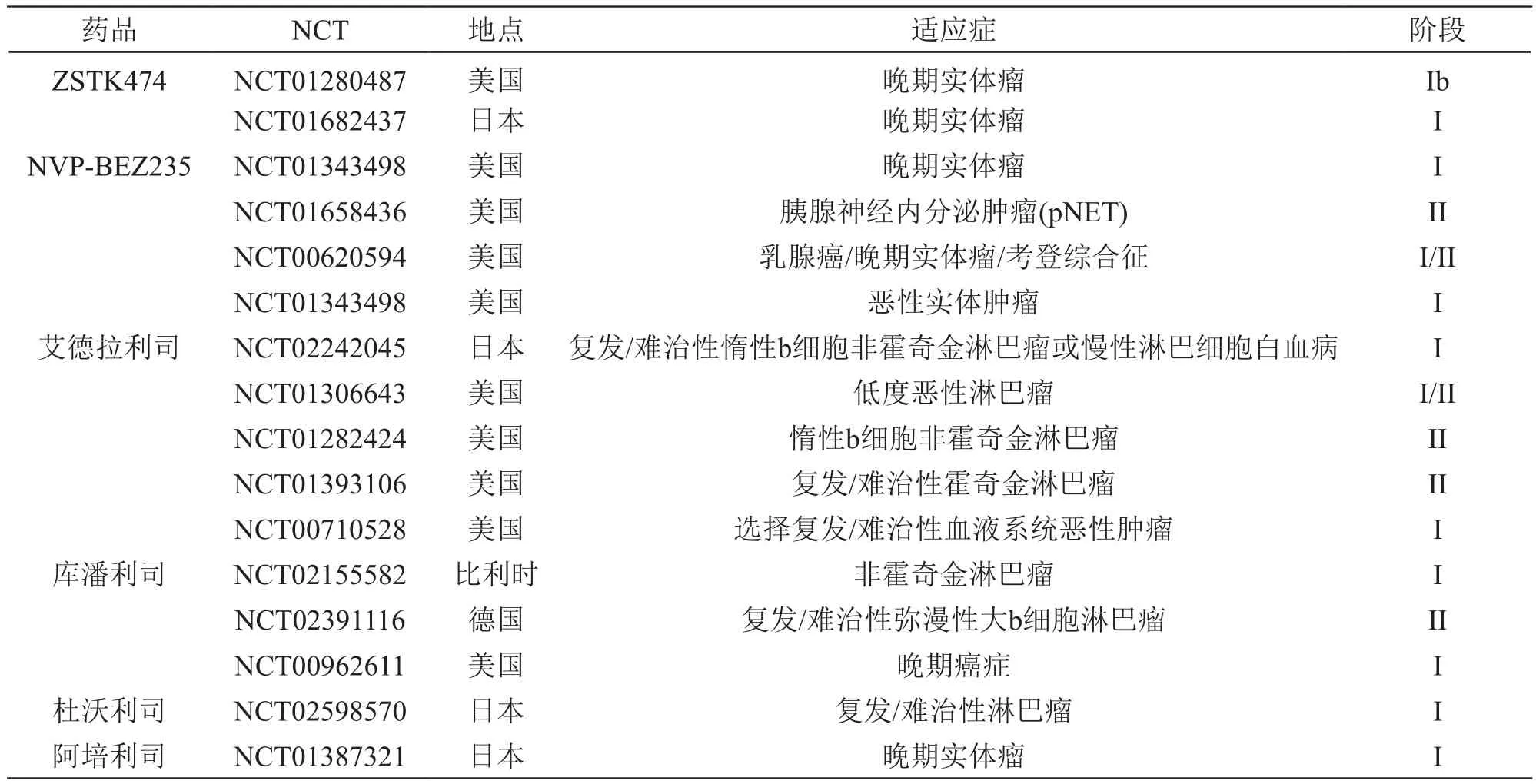
表2 具有代表性的PI3K抑制剂单独用药临床试验(已完结)
3.2 Idelalisib联合用药
前文已述,Idelalisib 已被批准用于复发/难治性慢性淋巴细胞白血病的治疗,伊马替尼也是该类疾病治疗的药物,但耐受性差及部分患者不敏感使伊马替尼的使用受到限制。研究表明Idelalisib 与伊马替尼联合应用在体外试验中,通过提高凋亡分子Bad、Bax表达等而达到协同诱导肿瘤细胞凋亡作用,且呈剂量依赖性[21]。另一项研究证实Idelalisib与化疗药物苯达莫司汀联合用药,通过诱导慢性淋巴细胞白血病细胞DNA损伤而引起细胞凋亡,产生协同的抗肿瘤活性[22]。此外,Idelalisib联合使用苯达莫司汀和利妥昔单抗治疗复发或难治性慢性淋巴细胞白血病已进入临床Ⅲ期试验,已有的结果证实相对于安慰剂组可减缓肿瘤病情进展,降低死亡风险[23]。而Idelalisib 与利妥昔单抗联合用药用于复发/难治性慢性淋巴细胞白血病老年患者已进入II期临床试验[24]。
3.3 Copanlisib联合用药
Seiichi等[25]报道Copanlisib 在体内和体外试验中都可抑制VEGF所介导的血管生成,与卡非佐米联用可增强卡非佐米的细胞毒性,诱导多发性骨髓瘤细胞的凋亡。Ye等[26]研究表明,Copanlisib与索拉非尼联合用药产生协同的抗肿瘤活性。Copanlisib通过对下调cyclin D1引起细胞周期阻滞,对索拉非尼耐药肝癌细胞系有较好的抗肿瘤活性,并能增强索拉非尼的促进肿瘤细胞凋亡作用。Kim等[27]研究表明,Copanlisib与吉西他滨联合用药,表现出良好的临床反应和药代动力学特征,安全性可控,该组合用于胆道癌现已进入II期临床试验。
3.4 Duvelisib联合用药
Flinn等[28]开展了一项Duvelisib+利妥昔单抗或Duvelisib+苯达莫司汀+利妥昔单抗联合用药的I期临床试验,以复发/难治性非霍奇金淋巴瘤或慢性淋巴细胞白血病患者为研究对象,目的在于评价联合用药方案的药物安全性,结果表明,该联合用药方案并未出现药物毒性增加,但该组合的具体临床疗效有待进一步研究。
3.5 Alpelisib联合用药
Alpelisib联合来曲唑用于晚期转移性乳腺癌的治疗已进入Ib期临床试验,结果表明,该联合用药方案药物毒性安全可控,并具有持续的治疗效果[29]。对于KRAS突变性非小细胞肺癌,有研究报道Alpelisib 与MEK抑制剂司美替尼在体内外试验中具有协同抗肿瘤作用,Alpelisib 提高了肿瘤细胞对司美替尼的敏感性,克服了肿瘤细胞对MEK抑制剂的耐药[30]。此外,Alpelisib和紫杉醇联合用于晚期实体瘤,Alpelisib和奥拉帕尼联合用于上皮性卵巢癌,Alpelisib和康奈非尼以及西妥昔单抗三药联合用于治疗转移性braf突变型结直肠癌均已进入Ib期临床试验[31-33]。
3.6 Buparlisib联合用药
Buparlisib(BKM120)是诺华制药研发的一种口服泛PI3K抑制剂,该化合物具有良好的血脑屏障穿透性,且BKM120具有良好的口服生物利用度,在脑内的浓度明显高于机体其他组织部位,因此BKM120被认为是颅内肿瘤靶向治疗的候选药物,目前BKM120对于颅内肿瘤的治疗已进入III期临床试验[34]。此外,BKM120与卡培他滨联合用于治疗转移性乳腺癌已进入I期临床试验,表现出良好的耐受性[35]。BKM120与贝伐单抗联合用于转移性肾细胞癌进入I期临床试验,结果显示该联合用药方案安全性可控,耐受性良好[36]。BKM120与奥拉帕尼联合用于高浆液性卵巢癌和乳腺癌也已进入I期临床试验,结果显示联合用药能产生协同的抗肿瘤作用,但毒副作用提示BKM120的剂量需降低[37]。此外,BKM120和MEK 1/2抑制剂比美替尼联合用于RAS/BRAF突变的晚期实体瘤患者,BKM120与卡铂和紫杉醇联合用于治疗PTEN缺失肿瘤患者均已进入 Ib期临床试验。但均未表现出令人满意的药物安全性[38-39]。BKM120可穿透血脑屏障,和贝伐单抗联合用药用于复发/难治性胶质母细胞瘤已进入Ⅱ期临床试验,但试验结果表明联合用药相对于贝伐单抗单独用药并没有表现出明显的疗效优势,尽管BKM120的剂量较低,却因为BKM120的加入引起严重的额外药物毒性,此外该联合用药方案用于难治性实体瘤进入I/II期临床试验[40]。BKM120与曲妥珠单抗和紫杉醇联合用于HER2+原发性乳腺癌同样因为耐受性和疗效问题在II期临床试验中被否定[41]。而BKM120与紫杉醇联合用药用于HER2-晚期乳腺癌的治疗因为临床效益问题终止于II期临床试验[42]。
3.7 ZSTK474联合用药
我们的研究证实ZSTK474通过抑制PI3K/AKT通路对人源慢性粒细胞白血病K562细胞及多药耐药细胞系K562/A02产生细胞增殖抑制和细胞周期G1期阻滞作用,当与伊马替尼联合用药时产生协同抗肿瘤活性[43]。而对于急性髓系白血病,ZSTK474对急性髓系白血病HL60细胞系及耐阿霉素细胞系HL60/ADR均产生细胞周期G1期阻滞和增殖抑制作用,当与阿糖胞苷或长春新碱联合用药时,会产生协同的抗肿瘤活性[44]。对于多形性胶质母细胞瘤,ZSTK474和蛋白酶体抑制剂Velcade联合用药促进肿瘤细胞凋亡,产生协同的抗肿瘤活性[45]。
3.8 其他
Umbralisib(TGR-1202)是TG治疗研发的一种新型的口服PI3Kδ抑制剂。Davids等[46]报道了Umbralisib与依鲁替尼联合用于治疗复发或难治性慢性淋巴细胞白血病和套细胞淋巴瘤的Ib期临床试验。研究结果显示该联合用药方案安全有效。此外Umbralisib联合依鲁替尼和优步利妥昔单抗用于复发或难治性B细胞恶性肿瘤也已进入I期临床试验。
Pictilisib,由基因泰克研发,是一种新型的口服I类PI3K抑制剂,与EGFR抑制剂厄洛替尼联合用于治疗晚期实体瘤进入I期临床试验,但抗肿瘤活性有限[47]。对于激素受体阳性、HER2阴性、局部复发或转移性乳腺癌,Pictilisib与紫杉醇联合用药进入II期临床试验,但该联合用药无进展生存期没有明显改变[48]。对于雌激素受体阳性乳腺癌患者,Pictilisib与阿那曲唑联合用药进入II期临床试验,该联合用药方案可显著增加抗肿瘤细胞增殖的活性[49]。Pictilisib与一线含铂化疗方案联合用于治疗晚期非小细胞肺癌进入Ib期临床试验,显示出可接受的安全性以及良好的抗肿瘤活性[50]。
Voxtalisib (SAR245409,XL765),由赛诺菲公司研发,是一种高选择性、强效的PI3K/mTOR双重抑制剂。与MEK抑制剂皮吗赛替联合用于治疗晚期实体瘤进入Ib期临床试验,但结果显示该联合用药方案临床抗肿瘤活性和长期耐受性均较差[51]。
PX-866,由昂克赛龙公司研发,是一种I类PI3K抑制剂。PX-866与维莫非尼联合用药用于晚期BRAF V600突变实体瘤进入I期临床试验,耐受性良好,最常见的不良反应为胃肠道反应[52]。
GSK2126458(GSK458),由葛兰素史克公司研发,是一种PI3K/mTOR双重抑制剂。GSK2126458与曲美替尼联合用于治疗晚期实体瘤进入Ib期临床试验。但该联合用药方案被证实临床抗肿瘤活性低,且耐受性差,因此已被中止[53]。
PF-04691502和Gedatolisib (PF-05212384) 均为辉瑞公司研发的PI3K/mTOR双重抑制剂。一项I期临床试验评估了PF-04691502和PF-05212384分别和伊立替康或MEK抑制剂PD-0325901联合用于治疗晚期实体瘤的安全性、药动学特征及初步的抗肿瘤活性。在试验过程中因为耐受性差以及抗肿瘤活性较低,PF-04691502的两组联合用药方案被迫中止。而PF-05212384两组具有较高的临床效益[54]。
4 小结与展望
本文在介绍了PI3K抑制剂的基础上,综述了其与其他疗法联合抗肿瘤的研究进展。十几年来肿瘤学家及药学家们对PI3K抑制剂的抗肿瘤疗效寄予了极大的希望,振奋人心的临床前研究结果也鼓励世界上各大制药企业纷纷投入了巨资进行该类药物的研发。但越来越多临床试验结果的出现使科学家们认识到PI3K抑制剂单独用药的抗肿瘤效果并没有像当初期待的那么好,这可能归咎于皮疹、腹泻等副作用的出现使得病人不得不停药或降低剂量,使得疗效受限。联合给药的方案可降低每种药物的剂量,从而减轻毒副作用。根据目前的临床试验结果看,一些联合用药方案取得了较好的疗效,而另一些方案好像并没有解决耐受性等问题,这可能与PI3K抑制剂所联合的药物种类或靶点有关。在联合用药的方案设计上还有许多工作可做,例如在有些情况下可探讨两种药物的序贯用药或脉冲给药。

表3 近3年具有代表性的进入临床试验/批准上市的PI3K抑制剂联合用药
已上市或开发中的PI3K抑制剂大致包括PI3K/mTOR双重抑制剂、泛PI3K抑制剂以及PI3K亚型特异性抑制剂。关于哪一类抑制剂更好的问题,目前似乎仍没有定论。最早进入临床试验的诺华制药研发的NVP-BEZ235曾以一石二鸟同时靶向PI3K和mTOR两个靶点而受到关注,但包括该药在内的许多PI3K/mTOR双重抑制剂的临床试验似乎已终止。已上市的药物中包括PI3Kδ亚型抑制剂Idelalisib、PI3Kα亚型抑制剂Alpelisib、同时靶向PI3Kδ和γ亚型的抑制剂Duvelisib以及泛PI3K抑制剂Copanlisib,因此至少从安全性的角度来看,泛PI3K抑制剂并没有因为其抑制所有4种PI3K亚型而产生不可耐受的副作用。以上4种上市药物,只有Alpelisib被用作实体瘤的治疗。但鉴于多种PI3K抑制剂正处于临床II期和III期的实体瘤治疗试验中,预计今后用于实体瘤治疗的新型PI3K抑制剂会陆续上市。
与老年性痴呆、艾滋病等其它难治性疾病一样,抗肿瘤药物的研发道路任重而道远,但随着肿瘤分子分型及标志物等精准医学研究的发展,以PI3K抑制剂为代表的分子靶向药物在肿瘤临床治疗中一定能发挥更大的作用。
猜你喜欢
传染病信息(2022年3期)2022-07-15 08:24:12
中老年保健(2021年5期)2021-08-24 07:06:38
中老年保健(2021年6期)2021-08-24 06:53:48
中老年保健(2021年11期)2021-08-22 03:14:10
中华养生保健(2020年3期)2020-11-16 00:53:14
基层中医药(2020年5期)2020-09-11 06:32:04
家庭医学(下半月)(2019年11期)2020-01-16 08:39:08
磁共振成像(2015年5期)2015-12-23 08:52:50
天津医科大学学报(2015年3期)2015-06-05 12:21:49
中国合理用药探索(2012年2期)2012-03-20 16:30:30
