
髓样分化因子88 多态性的研究进展
2020-05-27 09:51:36胡玉懿潘柏申
检验医学 2020年4期
胡玉懿,陈 朴 郭 玮,潘柏申
(复旦大学附属中山医院检验科,上海 200032)
人类拥有强大的自身免疫系统,用于防御外来病原微生物。人体的免疫系统可分为两大类:固有免疫和适应性免疫。原先以为人体内发挥主要免疫功能的是适应性免疫,但随着20世纪90年代Toll样受体(Toll-like receptor,TLR)的功能逐渐被揭示,固有免疫的作用也日益彰显。固有免疫是一种天然的防御功能,是人体抵御外界病原微生物的第一道防线,主要由吞噬细胞介导。固有免疫由能识别病原相关分子模式的受体介导,这些受体统称为模式识别受体。模式识别受体信号的转导可激活很多信号通路,从而调控各免疫应答表达的基因,启动固有免疫以及适应性免疫反应,以此来抵抗病原微生物的攻击。到目前为止,TLR是已发现的最重要的模式识别受体,多表达于有免疫功能的组织和细胞中[1],能选择性识别病原体所携带的相关分子,诱导树突状细胞及巨噬细胞活化和成熟,促进细胞因子分泌,进行免疫应答。目前已发现TLR家族13个成员,虽然不同的TLR识别的病原相关分子模式不同,但髓样分化因子88(myeloid differentiation factor 88,MYD88)是此类信号传导通路中重要的转接分子,在TLR信号通路中起传导信号的作用,激活下游多种转录因子,最终启动固有免疫应答[2]。本文对MYD88的结构、基本功能、在信号传导通路中的作用及MYD88 L265P基因突变与疾病的关联等进行综述。
1 MYD88的分子结构和功能
MYD88基因主要表达于免疫细胞中,如胸腺细胞、单核细胞、T细胞、B细胞、1型辅助T细胞(helper T cell 1,Th1)和2型辅助T细胞(helper T cell 2,Th2)。MYD88最初在髓样细胞的分化过程中被发现,是细胞质中的一种可溶性蛋白,由296个氨基酸残基组成,相对分子质量为3.5×104,是TLR/白细胞介素1受体(interleukin-1 receptor,IL-1R)家族和死亡结构域家族成员。除TLR3信号通路外,MYD88是所有TLR家族信号通路中的必需分子,因而被称为万能接头蛋白。
MYD88有3个功能结构域,分别是N端的死亡结构域、中端结构域和C端高度保守的Toll样受体/白细胞介素1受体(Toll-like receptor/interleukin 1 receptor,TIR)域。N端参与的信号通路与凋亡有关,通过招募、结合和活化其他携带死亡结构域的分子,如白细胞介素-1受体相关激酶(interleukin 1 receptor-associated kinase,IRAK),进一步传导相关信号[3]。C端携带与TLR分子一样的结构域,其氨基酸约有130个,与同型相互作用并与TLR/IL-1R相接后,可激活丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)和核因子-κB(nuclear factor kappa B,NF-κB)等通路,向细胞内传导信号[4]。
2 MYD88信号通路
TLR信号通路有MYD88和β-干扰素Toll样受体/白细胞介素1受体结构域衔接蛋白(Toll-like receptor/interleukin-1 receptor domain containing adaptor protein inducing interferon-β,TRIF)2条传导途径,MYD88依赖性途径在TLR信号传导通路中很多见,而TRIF依赖性途径则是TLR3和TLR4独有的[5]。该途径与β-干扰素(interferonbeta,IFN-β)的产生和树突状细胞(dendritic cell,DC)的成熟密切相连,可引起Ⅰ型干扰素反应,产生IFN-β,在信号传导中发挥关键作用[6]。
2.1 MYD88依赖的信号通路
除TLR3外,其他TLR及IL-1R家族都是依靠MYD88依赖性信号传导途径来传递信号的[7]。MYD88的下游信号通路大致相同,MYD88与TLR相互作用后,便可募集IRAK家族成员[8]。
在处于静息状态的细胞中,MYD88调节蛋白-Toll相关蛋白与IRAK1稳定结合。受配体刺激后,MYD88通过TIR-TIR的相互作用,被招募到激活的TLR的TIR区域;同时,MYD88利用其死亡结构域招募IRAK4,并使其发挥激酶作用,使IRAK1和IRAK2磷酸化,形成Myddosome复合物[9]。此刻,MYD88 N端的死亡结构域与IRAK-1、IRAK-4及肿瘤坏死因子受体相关因子(tumor necrosis factor receptor-associated factor,TRAF)6相连,其C端的TIR结构域与受体结合。IRAK-1和IRAK-2进一步发生磷酸化,随后通过TRAF结合域与E3泛素化酶、TRAF6结合[10]。TRAF6通过双重活化途径来传导信号。一条途径是MAPK途径,利用细胞外调节蛋白激酶(extracellular regulated protein kinase,ERK)、c-Jun氨基末端激酶(c-Jun N-terminal kinase,JUK)和p38,激活JUK家族的转录因子c-Jun和c-fos,使其进入细胞核,调控白细胞介素(interleukin,IL)-6、IL-1及肿瘤坏死因子α(tumor necrosis factor alpha,TNF-α)等炎症因子转录[9],相关过程见图1;另一条为激活NF-κB途径,静息时NF-κB二聚体隔离在细胞质上,当NF-κB被激活后,转化生长因子β活化激酶1发生磷酸化并激活核因子-κB抑制蛋白激酶(inhibitor of nuclear factor kappa-B kinase,IKK)复合物的活性,使核因子-κB抑制蛋白(inhibitor of nuclear factor kappa-B,IκB)磷酸化并降解蛋白酶体,退化的IκB释放NF-κB,调控炎症基因的转录[11]。
2.2 TRIF依懒的信号通路
MYD88非依赖的TLR3信号通路需要适配器分子TRIF来激活下游通路,包括干扰素调节因子3(interferon regulatory factor 3,IRF3)的激活和IFN-β的产生。TRIF也参与Ⅰ型干扰素反应中TLR4的下游信号传导。通过各种配体的结合以及TLR的TIR区与TIR区衔接蛋白(TIR domaincontaining adapter protein,TIRAP)、TRIF和TRIF相关衔接蛋白(TIRF-related adaptor molecule,TRAM)的相互作用,来激活NF-κB[12]。

图1 MYD88依赖、非依赖通路的下游信号传导[9]
3 MYD88介导TLR信号通路引发的下游效应和对细胞因子的调节
MYD88依赖通路是除TLR3外的其他TLR的共同通路,其重要性也显而易见,可以激活多种下游转录因子,刺激促炎细胞因子的形成,诱导多种防御蛋白的转录,促进TNF-α、IFN-β、IL-1β、IL-6、IL-12及IL-18的表达[13]。若敲除巨噬细胞的MYD88基因,其产生TNF-α的功能也会受损。
除激活NF-κB及MAPK通路外,在TLR-7和TLR-9被活化后,MYD88可以招募TRAF3来激活TBK1和IKKε,TBK1和IKKε可使干扰素调节因子-7(interferon regulatory factor 7,IRF7)磷酸化,产生α-干扰素(interferon-alpha,IFN-α),在抗病毒反应中起关键作用[9]。
TLR/MYD88信号通路是一条具有多种调节功能的信号传导通路,在免疫反应、炎症反应及肿瘤的发生、发展过程中均具有重要作用。
4 MYD88基因L265P突变
MYD88基因L265P突变是MYD88编码序列第794位碱基发生T→C突变,使得MYD88蛋白编码区第265位的亮氨酸错义为脯氨酸。高频发的体细胞MYD88 L265P突变通过配对肿瘤/正常全基因组测序在华氏巨球蛋白血症(Waldenstrom's macroglobulinemia,WM)患者中被发现后,又被Sanger测序和等位基因特异性-聚合酶链反应(allele specific polymerase chain reaction,AS-PCR)证实[14]。
有研究者在使用更为敏感的AS-PCR检测CD19磁珠分选或未分选的骨髓细胞时发现,90%~100%的WM患者有MYD88 L265P突变;而骨髓瘤,包括IgM骨髓瘤患者却很少有MYD88 L265P突变;在少部分(6.5%)边缘区域淋巴瘤患者中,该突变也很少,而这类患者有很多与WM患者相似的临床特征[15]。
5 MYD88 L265P突变与肿瘤的关系
5.1 淋巴细胞相关肿瘤中MYD88 L265P的突变情况
JARDIN[16]发现,在多种B细胞肿瘤患者和3%的慢性淋巴细胞白血病患者中发现MYD88 L265P突变。该突变会异常激活信号通路,而异常的MYD88可能与B细胞肿瘤的发生、发展及预后有关。
5.1.1 淋巴浆细胞淋巴瘤(lymphoplasmacytoid lymphoma,LPL)LPL是一种惰性成熟小B细胞淋巴瘤,约占非霍奇金淋巴瘤(non-Hodgkin's lymphoma,NHL)的2%,非常罕见。LPL的病理特点为小淋巴细胞、浆细胞样淋巴细胞,大多数侵犯骨髓,可浸润到淋巴结和脾脏。当LPL侵犯骨髓同时伴有血清单克隆性IgM丙种球蛋白时即可诊断为WM[17]。WM占LPL的90%~95%,仅少数LPL患者分泌单克隆性IgA、IgG或不分泌单克隆性免疫球蛋白[18]。临床上常需要与伴有IgM型单克隆丙种球蛋白血症的IgM型多发性骨髓瘤、慢性淋巴细胞白血病以及IgM相关疾病等鉴别,而IgM型意义未明的单克隆丙种球蛋白病(monoclonal gammopathy of undetermined significance,MGUS)易转化发展成WM[19]。TREON等[14]对CD19磁珠分选后的WM患者骨髓细胞的全基因组测序结果显示,有90%的患者染色体3p22.2位点38182641有体细胞突变(T→C),即MYD88 L265P突变。另有研究发现MYD88 L265P突变可激活IRAK介导的NF-κB信号通路,促进细胞增殖[20]。提示MYD88 L265P突变是LPL中具有高检出率且特异的突变,可用于LPL与其他B细胞肿瘤的鉴别诊断。GACHARD等[21]在WM患者中检测出了MYD88 L265P突变,突变率为70%~100%。在IgM型MGUS患者中,MYD88 L265P突变的阳性检出率远低于WM患者,因此其可能是IgM型MGUS恶性转化为WM基因层面的推动原因。XU等[17]采用敏感性和特异性更高的AS-PCR进行定性、定量检测,结果显示MYD88 L265P突变表达量越高,骨髓受累程度越重,IgM水平越高,IgA、IgG水平降低的程度亦越严重;如果治疗有效,则MYD88 L265P突变的表达量也会随之降低甚至转为阴性。因此,在IgM型MGUS患者中,MYD88 L265P突变也较常见,MYD88 L265P表达量越高,越易向WM转化。总之,MYD88 L265P突变与LPL的临床特征及预后相关,可用于评估LPL的疗效。
5.1.2 弥漫大B细胞淋巴瘤(diffuse large B-cell lymphoma,DLBCL)DLBCL是NHL最常见的一种类型,发病率占NHL的30%~40%[22]。在我国,DLBCL占NHL的45.8%,占所有淋巴瘤的40.1%[23]。依据基因表达的不同,可将DLBCL分为生发中心B细胞样淋巴瘤、活化B细胞样淋巴瘤(activated B-cell type,ABC)和原发纵膈弥漫大B细胞淋巴瘤(primary mediastinal large B-cell lymphoma,PMBL)。ABC的特点是过度激活的NF-κB信号通路。NGO等[15]的研究结果显示,约有29%的ABC存在MYD88 L265P突变,而其他类型的DLBCL(伯基特淋巴瘤和生发中心B细胞样淋巴瘤)则罕见该突变。ROVIRA等[24]的研究证实了该结论。MYD88会产生活性蛋白,MYD88 L265P过度表达可促进DLBCL细胞的选择性增殖,而恶性细胞的增殖常与IRAK1、IRAK4活性增强及下游NF-κB信号通路激活有关。在内源性表达MYD88 L265P的WM细胞系中加入MYD88、IRAK1或IRAK4的抑制剂,均会降低NF-κB p65的核染色,证实MYD88 L265P是通过NF-κB来传导信号的[14]。除NF-κB外,敲除MYD88或使用IRAK1、IRAK4抑制剂均可在MYD88过度表达的DLBCL细胞系中通过信号转导子和转录激活子(signal transducer and activator of transcription3,STAT3)减少IL-6和IL-10的自分泌,说明MYD88 L265P还可以调节和激活酪氨酸激酶(janus kinase,JAK)-STAT3信号途径,促进肿瘤形成[25-26]。阻断STAT3或IL-1可明显增加MYD88 L265P突变淋巴细胞对CD8+T细胞介导的细胞毒性的敏感性[27]。此外,MYD88介导的TLR信号通路还可直接影响适应性免疫反应。信号通路和NF-κB的异常激活会产生和释放肿瘤微环境中一系列炎性因子和细胞因子,导致免疫抑制,促进肿瘤免疫逃逸,相关机制见图2[9]。
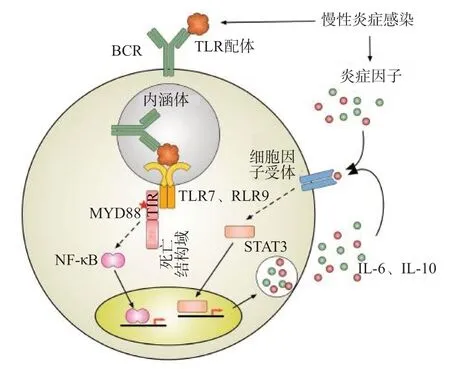
图2 B细胞的活化和炎症[9]
5.1.3 其他B细胞肿瘤 有研究结果显示,在33%的中枢神经系统淋巴瘤、9%的黏膜相关淋巴瘤、2.9%的慢性淋巴细胞白血病患者中发现了MYD88 L265P突变,但MYD88 L265P突变对这些疾病的诊疗价值尚不完全明确[28]。
5.2 肝细胞肝癌(hepatocellular carcinoma,HCC)中MYD88 L265P的突变情况
近年来,HCC的发病率和死亡率呈上升趋势[29]。HCC患者大多有不同程度的肝炎病史,乙型肝炎病毒(hepatitis B virus,HBV)感染和炎症促进了HCC的发生、发展。
TLR能够识别病原相关分子模式和损伤相关分子模式(damage-associated molecular pattern,DAMP)[30]。TLR4高表达于白血病、肝癌、乳腺癌、肺癌、结直肠癌组织中。TLR识别配体后,可传递给细胞内的MYD88、MYD88接头样蛋白、TLR相关的干扰素活化因子和TLR相关的分子。IL-17A和IL-23是炎症的关键推动因子,可导致肿瘤的发生,并与HCC中的TLR4有关[31]。在HCC早期,持续激活的TLR4信号可使HBV感染导致的慢性炎症迁延不愈,加上乙型肝炎表面抗原可激活NF-κB途径,从而使慢性乙型肝炎向恶性肿瘤方向进展。另外,MYD88还可引起免疫抑制,增加癌细胞逃逸的可能性。因此,MYD88在肝细胞癌变过程中起促进作用[29]。
5.3 乳腺癌中MYD88 L265P的突变情况
乳腺癌居妇科恶性肿瘤患者死亡的第1位,90%以上的致死原因是肿瘤转移或复发。持续难愈的炎症可诱发癌症。慢性炎症可刺激肿瘤组织释放能促进其生长的因子,反过来又形成炎症微环境。长此以往,该恶性循环会诱导肿瘤的发生、发展。有研究结果显示,乳腺癌细胞株高表达TLR9,其表达量与肿瘤病理分期分级、转移和预后密切相关[32];TLR2和TLR4与乳腺癌亦有关联[33]。TLR4和MYD88的蛋白表达与不良临床特征显著相关[34]。
5.4 其他肿瘤中MYD88 L265P的突变情况
固有免疫引导的炎症反应主要由TLR介导,而TLR4是第1个被发现的、与肿瘤关系较为密切的TLR相关蛋白,其活化的信号转导通路属于经典的依赖MYD88的信号转导通路。由TLR诱导的信号转导通路在肿瘤,特别是与炎症相关的肿瘤,如肝癌、结肠癌[35]、胃癌、食管癌[36]、胰腺癌、肺癌、卵巢癌和宫颈癌等的发生、发展中起重要作用。有研究结果显示,TLR4和MYD88在所有大肠癌组织中均有表达,TLR4和MYD88高表达与肝脏转移有关,并且是大肠癌患者预后不佳的一个独立危险因素[37]。胃癌、胃溃疡和慢性浅表性胃炎中也均有MYD88的阳性表达。TLR4和TLR9在肺癌组织中的表达也明显高于癌旁组织,且肿瘤细胞分化的恶性程度越高,TLR4的表达也越高。此外,TLR4在食管癌中的表达水平亦很高,其表达与食管癌的分化程度呈负相关(r=-0.522,P<0.05)[38]。
6 其他疾病中MYD88 L265P的突变情况
6.1 炎症性肠病(inflammatory bowel disease,IBD)
IBD是发病原因不明的一种疾病,可能源自免疫功能失调,会有持续的炎症反应。HOSHI等[39]比较了IL-10敲除的小鼠和野生型小鼠发生IBD的情况,发现缺乏IL-10表达时,肠道细菌可以利用单核吞噬细胞中的MYD88依赖通路,诱发IBD。ZHOU等[40]的研究结果显示,肠道的炎症受到TLR4-MYD88-MAPK信号传导和NF-κB途径的调节,而且哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)依赖性自噬的调节机制可为IBD提供潜在的治疗靶点。
6.2 心血管疾病
MYD88依赖性NF-κB信号途径可导致心肌肥大,进而阻断其信号通路的传导,可有效降低心肌肥大的发生率。有研究结果显示,缺少MYD88的小鼠发生早期动脉粥样硬化的概率显著降低[41]。MYD88在心肌梗死后可导致心肌肥厚和炎症的发展,其缺失对心脏功能改善、全身炎症情况缓解以及死亡率的降低均可能发挥正向作用[42]。
6.3 新生儿感染
张金萍等[43]的研究结果显示,MYD88 mRNA高表达于发生感染的新生儿中,表明MYD88依赖信号传导通路在新生儿的免疫系统中起关键作用。虽然降低胎儿体内MYD88的表达可以缓解炎症反应,但为了适应宫外复杂的病原体环境,尽量减少感染,新生儿需要快速获得固有免疫和适应性免疫。
7 MYD88 在相关疾病治疗中的应用
MYD88是TLR信号通路中举足轻重的分子,而TLR信号通路在先天性免疫反应和获得性免疫反应中均扮演着至关重要的角色。在很多疾病中都可发现TLR信号通路被激活,且其中的接头蛋白MYD88与肿瘤、感染性疾病、自身免疫性疾病以及炎性疾病等都有较为明确的关系。因此,MYD88也是治疗上述疾病的重要靶点。目前可利用基因敲除、RNA干扰来调节MYD88的表达,还可以将免疫检查点抑制剂与L265P信号传导网络组合在一起,形成靶向化学疗法[44]。
另外,增强MYD88信号传导通路也具有较大的潜在应用价值,如使用L265P衍生肽疫苗接种,在接种疫苗时可以通过活化MYD88信号通路,使接种患者获得更为有效的免疫力,还可提升疫苗的效能等[45]。因此,对某些疾病而言,MYD88激动剂可能是有益处的。但对于MYD88表达过强的患者,则易诱发自身免疫性疾病。MYD88还是当前治疗脓毒症的靶点,调控其信号通路的传导,可减弱脓毒症的炎性反应。MYD88缺失能抑制IBD、脑部创伤后的脑炎及心肌炎的发展,保护机体。但若彻底阻断该通路,则可能损伤免疫系统的防御功能[46]。
8 展望
MYD88是TLR信号传导通路中一个举足轻重的因子,是多种TLR信号传导的关键枢纽。目前,MYD88的功能及其与各种疾病的关系正逐步被揭示。MYD88 L265P突变在稳定MYD88-TIR结构域同源二聚体的同时也限制了其动态性,促进了细胞恶性增殖、逃避细胞周期抑制、阻滞细胞分化成熟,导致肿瘤形成。MYD88依赖通路在多种疾病中发挥关键作用,被认为是治疗这些疾病的重要靶点。但MYD88也具有致炎、促癌的作用。只有恰到好处地加以利用,才能更好地预防和治疗相关疾病。
猜你喜欢
传染病信息(2022年3期)2022-07-15 08:24:12
家庭医学(下半月)(2019年11期)2020-01-16 08:39:08
小学科学(学生版)(2019年10期)2019-11-16 08:55:04
广州大学学报(自然科学版)(2019年1期)2019-05-07 01:33:26
中国环境监察(2017年5期)2017-10-23 05:26:48
电测与仪表(2016年14期)2016-04-11 12:34:22
天津科技大学学报(2016年1期)2016-02-28 16:59:45
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10 08:41:53
磁共振成像(2015年5期)2015-12-23 08:52:50
天津医科大学学报(2015年3期)2015-06-05 12:21:49
