
Na修饰正四面体Si4团簇的结构及储氢性能研究*
2020-05-27 13:10:30阮文宋红莲伍冬兰罗文浪谢安东赵芳杰甘雅玲
中山大学学报(自然科学版)(中英文) 2020年3期
阮文,宋红莲,伍冬兰,罗文浪,谢安东,赵芳杰,甘雅玲
(井冈山大学数理学院,江西 吉安 343009)
当今世界能源消耗以石化能源为主, 但石化能源不可再生且在慢慢枯竭。石化能源消耗的同时,还释放出大量废气和有害气体,对环境造成了严重的污染和破坏。因此, 发展清洁、可再生的新能源成为各国的重要课题。众所周知,氢具有高燃烧值、能量密度高、环境友好、储量丰富、来源广泛、易于低成本获取等优点,被公认是一种前景良好的能源[1]。我国《国家能源发展战略行动计划(2014-2020)》将研究和开发储氢材料作为推进能源科技创新的重点方向之一;美国能源部则为储氢材料的发展制定了一系列的年度计划[2], 如设定2017年储氢质量分数的目标为5.5% 。但是, 氢的储存是其走向实用化和规模化的“瓶颈”。传统的储氢方式(高压氢气瓶和液氢储存)并不适合商业化使用, 储氢材料的开发可以较好地解决这一难题[3]。 研究表明:良好的储氢材料必须使材料和氢分子之间的相互作用介于物理吸附和化学吸附之间, 从而保证常温常压下具有较好的吸附氢行为[4-5]。为寻找理想的储氢材料, 世界各国对此进行了广泛研究,吸附储氢是近年来研究比较系统的方法。这种方法的典型代表为金属原子修饰碳或硼基纳米结构, 如:过渡金属Ti修饰碳纳米管[6]、Sc掺杂硼富勒烯材料[7]、碱土金属Ca原子修饰石墨烯[8]、碱金属修饰碳和硼团簇材料等[9-11]。相关研究也证实了碱金属由于原子之间的内聚能较小、原子质量相对较轻是理想的掺杂元素之一,如:碱金属修饰单壁碳纳米管[12]、多壁碳纳米管[13]、一维碳纳米材料等[14]。
目前,吸附储氢材料的理论研究大多仅限于以硼、碳为基础的纳米管或富勒烯,种类非常有限。考虑到硅与碳同族,物理化学性质与碳有许多相似性,也可形成纳米管和富勒烯等结构,且硅原子组成的纳米结构表面存在大量悬挂键,采用金属原子修饰硅富勒烯或其它纳米结构,有利于氢分子在其表面吸附。更为重要的是,硅的储量在地壳中排第二,仅次于氧,含量非常丰富,特别是在江西井冈山地区已探明具有储量非常丰富的硅矿源,便于提取并制备硅基纳米团簇材料。最近,Wang等[15]采用K原子修饰平面硅富勒烯,对其储氢性能进行了理论研究, 储氢质量分数达6.13%(w)。 阮文等采用密度泛函理论方法研究了锂修饰硅原子链的结构以及其与氢分子的相互作用[16], 分析了锂修饰Si原子链的储氢性能; 并对锂、钠原子修饰笼型Si5、Si6团簇的储氢性能进行了理论研究[17-19], 获得了碱金属修饰Si5、Si6团簇作为储氢材料的可能性。因此,硅除具有半导体方面的应用,还有作为储氢材料应用于氢能储存的可能性。
最近,Edison等[20]研究了碱金属Li修饰Si4团簇的结构特性, 指出正四面体的Si4Li4为最稳定的团簇结构。Sudip等[21]研究了碱金属Li修饰Si4团簇的电子特性及储氢性能, 表明其储氢质量分数达14.4 %。但,碱金属Na修饰Si4团簇可否作为储氢材料,目前还并不清楚。本文采用明尼苏达密度泛函M06方法系统研究了Na修饰正四面体Si4团簇的结构,及其与H2的相互作用, 并分析了Na修饰Si4作为储氢材料的可行性,为进一步研究Si基团簇材料的储氢性能及储氢机理提供了一定的理论基础。
1 研究方法
氢分子在材料表面吸附时, 通常与材料表面的结合为非成键的弱相互作用。这种弱相互作用在理论计算中往往容易被高估或低估, 一般考虑采用量子力学微扰法进行理论计算,但所耗计算时间巨大。明尼苏达密度泛函M06方法一个显著的优点是在拟合参数时就引入了弱相互作用体系,在非键相互作用、分子结构、振动频率和中程相关能量等方面都具有较高的预测精度,可以有效地解决分子间的非成键弱相互作用,比一般的泛函性能好很多,往往能得到与微扰理论相一致的结果,但计算所耗时间比微扰法少,已被很多主流量化程序采纳[22- 23]。故,本文采用M06/6-311++G (d, p)方法研究Si4团簇以及碱金属Na修饰正四面体Si4团簇的结构及其储氢性能。
计算过程中,结构优化以梯度、能量和位移是否收敛为标准, 优化结构的稳定性根据振动频率来判断。当出现虚频时,用消虚频程序进行结构调节,并重新进行优化,直至振动频率无虚频,得到体系的稳定结构。所有计算均在Gaussian 09程序包下完成,氢分子的平均吸附能(Ead)由下式计算[24]
Ead=[E(Na4Si4)+nE(H2)
-E(Na4Si4·nH2)]/n
(1)
式中,E(Na4Si4)、E(H2)和E(Na4Si4·nH2)分别表示Na4Si4、氢分子和Na4Si4·nH2团簇的基态能量,n为吸附的氢分子数目。
Na4Si4·nH2稳定结构的储氢质量分数可用(2)式计算,即
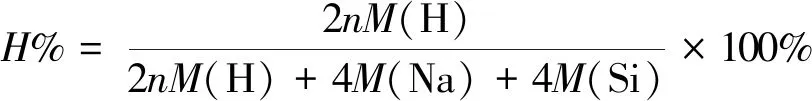
(2)
其中,M(H)、M(Na)和M(Si)分别为氢原子、钠原子和硅原子的相对原子质量。
2 计算结果
首先,对Si4可能存在的结构进行优化, 得到了稳定的正四面体结构(如图1中(a));接着, 研究H2直接在正四面体Si4团簇表面直接吸附,结果如图1中(b)-(d)。然后,考虑Na原子在正四面体Si4团簇结构上的各种修饰情况。Na原子可以在正四面体Si4团簇的四个面上吸附, 形成面位吸附Na4Si4正四面体(如图2中(a));也可以在正四面体Si4的四个原子上顶位吸附, 形成顶位吸附Na4Si4正四面体(如图2中(b));还可以在六个Si—Si键上以桥位吸附形成Na6Si4结构(如图2中(c))。最后, 研究面位吸附结构对H2分子的吸附情形,对于H2分子的吸附分别考虑不同分子数量、吸附的位置与方向进行初始化构型设计,采用同样的理论方法得到优化后的稳定吸附体Na4Si4·nH2(n=4-24)结构(如图3中(a)-(f)),并对Na4Si4吸附氢分子的物理化学性质做了理论研究,包括研究吸氢前后团簇体系的结构和能量变化、电荷分布与转移、H2的平均吸附能以及储氢质量分数等,Na原子修饰正四面体Si4团簇结构及Na4Si4·nH2(n=4-24)结构的物理化学性质相关计算结果分别列于表(1)-(3)。
3 结果分析与讨论
采用M06/6-311++G (d, p)方法优化得到正四面体Si4团簇的结构,如图1(a)所示。其Si—Si键长为0.246 8 nm, 频率计算不存在虚频,前线轨道能隙为2.032 eV,原子化能为3.395 eV,表明正四面体Si4团簇是势能面上的一个稳定结构。以此作为研究基础,直接在正四面体Si4团簇表面吸附H2,如图1(b)-1(c)。可见,单个H2在Si4团簇表面直接吸附时, 存在两种情况:一种是H2分子断键,以原子的形式吸附在Si4团簇表面,H—Si平均键长为0.169 4 nm,如图1(b),氢分子的吸附能为2.211 eV,此结合力太强,为典型的化学吸附,不利于氢的解离利用;另一种是以分子形式吸附,但Si4团簇的结构严重变形,几乎成为平面结构,此时H2与最近邻的Si原子之间距离为0.355 9 nm,氢分子的吸附能为1.539 eV,仍为化学吸附,也不利于氢的解离利用。多个H2在Si4团簇表面直接吸附时,氢虽然以分子形式吸附,但Si4团簇的结构严重变形,如图1(d),H2与最近邻的Si原子之间平均距离太远(约为0.42 nm),且氢分子的平均吸附能太小(为3.837 kJ·mol-1), 为典型的物理吸附, 难于有效吸附储氢。
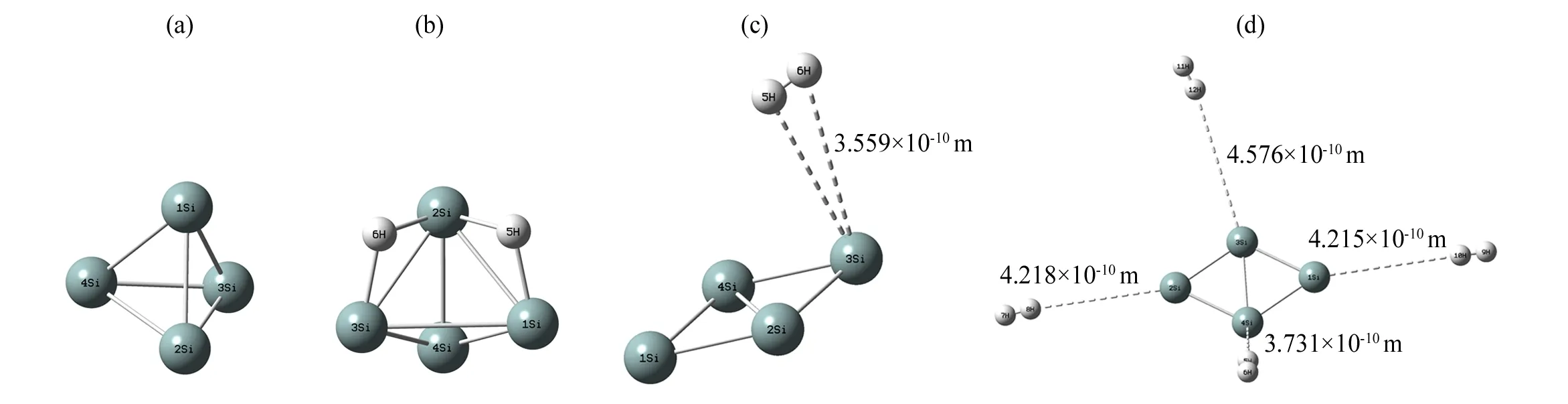
图1 Si4及Si4表面直接吸附氢分子的稳定结构Fig.1 Stable structures of Si4 and H2Si4 clusters
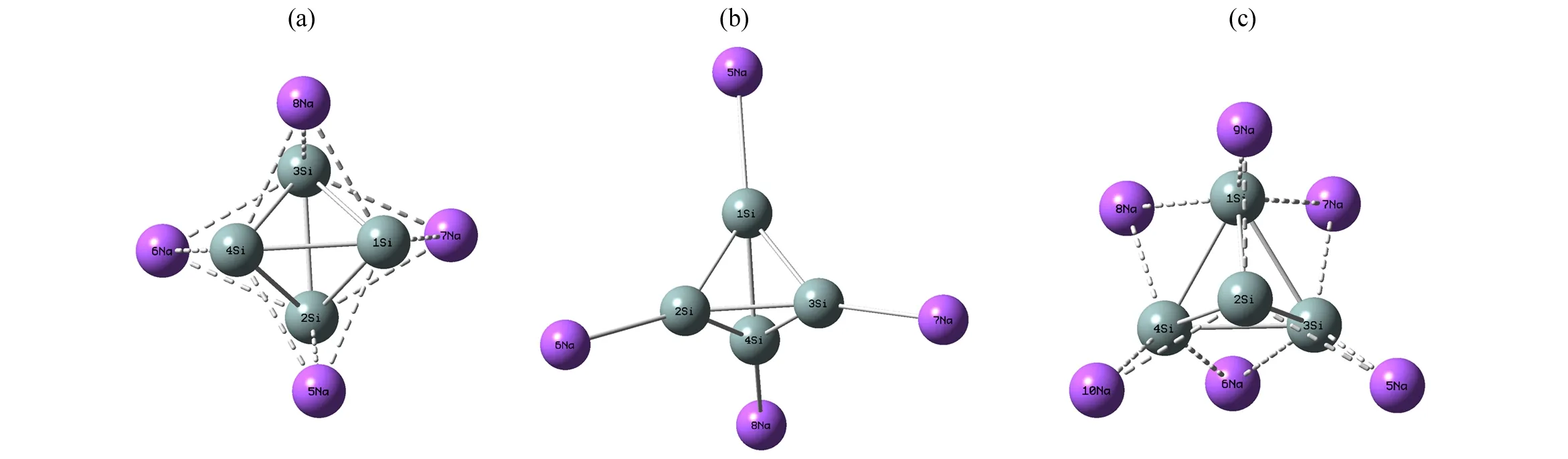
图2 Na4Si4、Na6Si4团簇的稳定结构Fig.2 Stable structures of Na4Si4 and Na6Si4 clusters
采用钠原子对其表面进行修饰时,分别对顶位吸附、桥位吸附以及面位吸附三种修饰情况进行了结构优化和频率计算,并计算了电离势(I)、电子亲和能(A)、前线轨道能隙(Eg)以及电负性(χ)、硬度(η)和最小谐振频率(ν),计算结果如表1。表1中钠原子面位吸附时电离势、能隙以及电负性和硬度最大, 电子亲和能最小, 且频率计算无虚频;顶位吸附时总能量不仅高于面吸附, 且频率计算存在一个虚频, 故顶位吸附不是稳定的结构, 而是一个过渡态,表明面位吸附结构最稳定。可见钠原子优先趋于吸附正四面体Si4团簇的四个表面上方构成Na4Si4团簇(如图2(a)), 此结构中Si—Na平均键长为0.283 9 nm,Si—Si平均键长为0.246 3 nm, 前线轨道能隙为2.408 eV。Majumder等[25]曾研究了Na4Si4团簇的各种结构,指出最低能量结构为面位吸附的正四面体构型,其计算的Si—Na平均键长为0.285 nm, Si—Si平均键长为0.249 nm, 前线轨道能隙为2.39 eV,与本文的理论计算结果基本一致。
如表2所示,自然电荷分析发现:每个钠原子失电荷0.763 e, 每个硅原子得电荷0.763 e, 表明该体系中钠原子与Si4正四面体间以部分离子键和部分共价键的形式相结合, 这不仅未使Si4正四面体结构发生明显改变, 而且体系的前线轨道能隙比未修饰的Si4能隙大,说明修饰后动力学稳定性显著增强。更有利的是,计算体系中Na原子的平均结合能为2.346 eV,远大于钠金属材料中原子的内聚能1.113 eV[26],表明Na原子在Si4表面不发生聚合,故面位修饰的Na4Si4体系可作为一个稳定的基体来实现对氢分子的吸附。
氢分子在Na4Si4团簇表面吸附的稳定结构如图3所示。图3中, 在每个Na原子周围添加一个、两个、三个……,依次递增至七个H2, 并利用M06/6-311++G(d, p)方法进行结构优化和频率计算, 发现:每个Na原子周围能够稳定吸附6个H2, 如图3(f)所示。当吸附至7个H2时,则有多个H2被排斥出去,使之与最临近的Na间距在0.350 nm以上(结构图未示出),表明其与Na4Si4相互作用太弱,不足于被Na4Si4体系有效吸附,这说明该体系吸附H2具有一定的饱和性。
由表2可见,Na—Si平均键长随吸附氢分子数目增多稍有增大,Na与H2平均距离随吸附氢分子数目增大而稍有减小,氢分子平均吸附能范围为3.926~9.927 kJ·mol-1, 介于物理吸附和化学解离吸附之间。在每个Na原子周围吸附1~6个H2时,氢分子的平均吸附能随所吸附氢分子数目的增多而增大。 这可以从自然电荷分布来加以分析,表2中随着吸附氢分子的增加,氢分子失电子数有少量增加,随即转移到Na4Si4体系上电子数增大,导致Na4Si4与氢分子之间的静电相互作用增大,从而吸附能也随之增大。当吸附到7个H2时相互作用显著变弱,这是由于Na4Si4表面周围空间有限,氢分子的增加导致空间出现位阻效应,再次说明体系吸氢具有一定的饱和性。

表1 Na在正四面体Si4团簇表面不同吸附方式下的电离势(I)、电子亲和能(A)、 能隙(Eg)、电负性(χ)、硬度(η)和最低谐振频率(ν)Table 1 Ionization potential(I), electronic affinity (A), energy gap (Eg), electronegativity (χ), hardness (η),and the lowest vibrational frequency (ν) of the different adsorption modes of the Na atoms decorated tetrahedron Si4 clusters
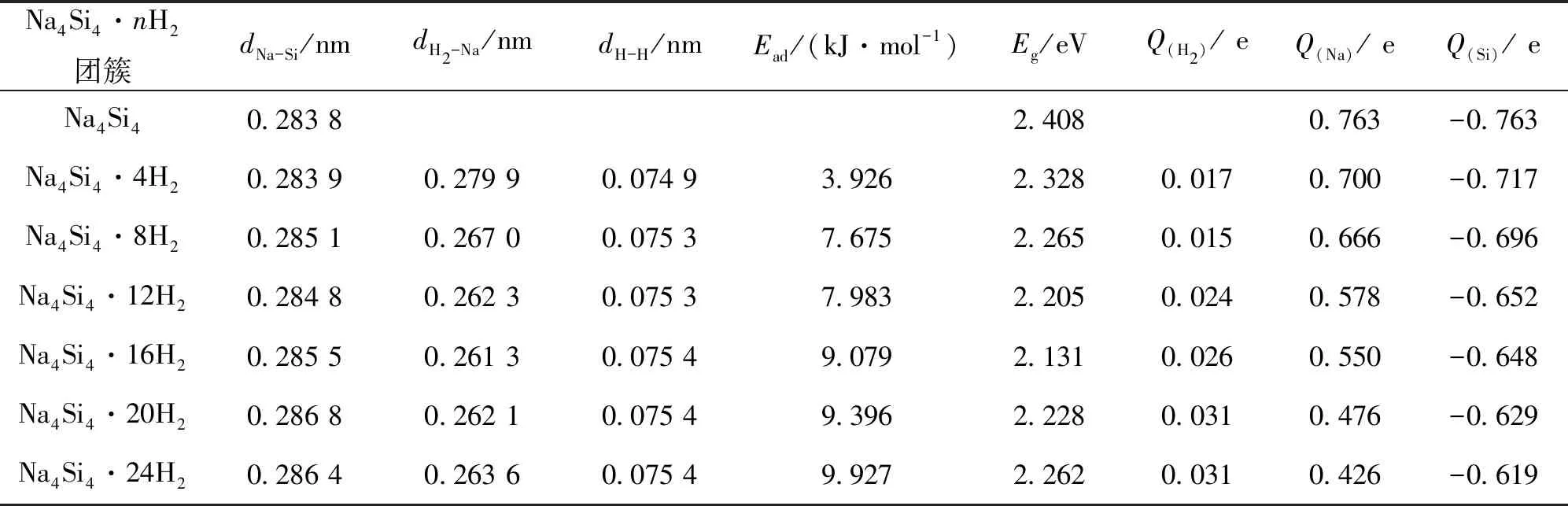
表2 Na4Si4·nH2团簇的平均键长和氢分子平均吸附能以及自然电荷分布Table 2 Average bond lengths of Na-Si (dNa—Si), H2-Na (dH2—Na )and H-H (dH—H ), average adsorption energies of H2 (Ead), energy gap(Eg) and the average natural charges on H2(Q(H2)), Na (Q(Na))and Si(Q(Si)) of the Na4Si4·nH2 clusters
用相同的方法计算得到孤立 H2分子的键长为0.074 7 nm。表2 中, 吸附在Na4Si4团簇表面后,H2分子的键长逐渐增大,变化范围为0.074 9~0.075 4 nm。其原因是氢分子靠近Na4Si4团簇时, 氢分子部分电荷转移到Na4Si4体系上, 失去少量电子后使氢分子中成键轨道之间的相互作用减弱而反键轨道相互作用相对加强,导致被吸附后H2分子键长比孤立H2分子键长稍长,但又不至于断裂成原子形式。随着所吸附的氢分子的增加,键长逐渐增大的原因是由于氢分子转移到Na4Si4体系上的电子数量有逐渐增加,氢分子中成键轨道之间的相互作用逐渐减小而反键轨道相互作用进一步加强所致。同样,由于更多的电荷发生转移导致Na4Si4体系与氢分子之间的相互作用随吸附的氢分子增加而增大,使得Na与H2的平均距离随吸附氢分子的数目增大而稍有减小。
自然电荷分布表明,Na4Si4团簇体系中,钠原子带正电荷,硅原子带负电荷,体系在空间形成静电场,氢分子接近该体系时,被静电场极化形成电偶极子,通过静电—偶极相互作用吸附在钠原子周围;并且在吸氢过程中有少量电子从氢分子转移到Na4Si4团簇体系,所以氢分子与Na4Si4团簇体系之间还存在较小的离子相互作用。随吸附的氢分子的增加,氢分子失电子数量稍有增加,从而转移到Na4Si4体系上的电子数量也增大,离子相互作用也稍微增大,使得Na4Si4体系与氢分子之间的相互作用稍有增大。但两种相互作用比较,Na4Si4体系的储氢机理主要是静电—偶极相互作用使得多个氢分子以介于物理吸附和化学解离吸附之间的方式吸附在Na4Si4的周围。利用(2)式计算得到Na4Si4·24H2的储氢质量分数理论值为19.15% 。
为研究该储氢体系的热力学性质,以Na4Si4·24H2为例计算体系吉布斯自由能相关的平均吸附能随温度的变化,计算公式如(3)式[27]。
ΔEG= [EG(Na4Si4)+24EG(H2)
-EG(Na4Si4·24H2)]/24
(3)
式中,EG(Na4Si4)、EG(H2)和EG(Na4Si4·24H2)分别表示Na4Si4、H2和Na4Si4·24H2团簇的吉布斯相关能。表3为不同温度下Na4Si4·24H2体系吉布斯自由能相关的平均吸附能,其随温度的变化趋势如图4所示。

表3 Na4Si4·24H2体系吉布斯自由能相关的平均吸附能随温度的变化Table 3 The temperature dependence of average adsorption energies with Gibbs free energy correction of H2 (ΔEG) of the Na4Si4·24H2 clusters
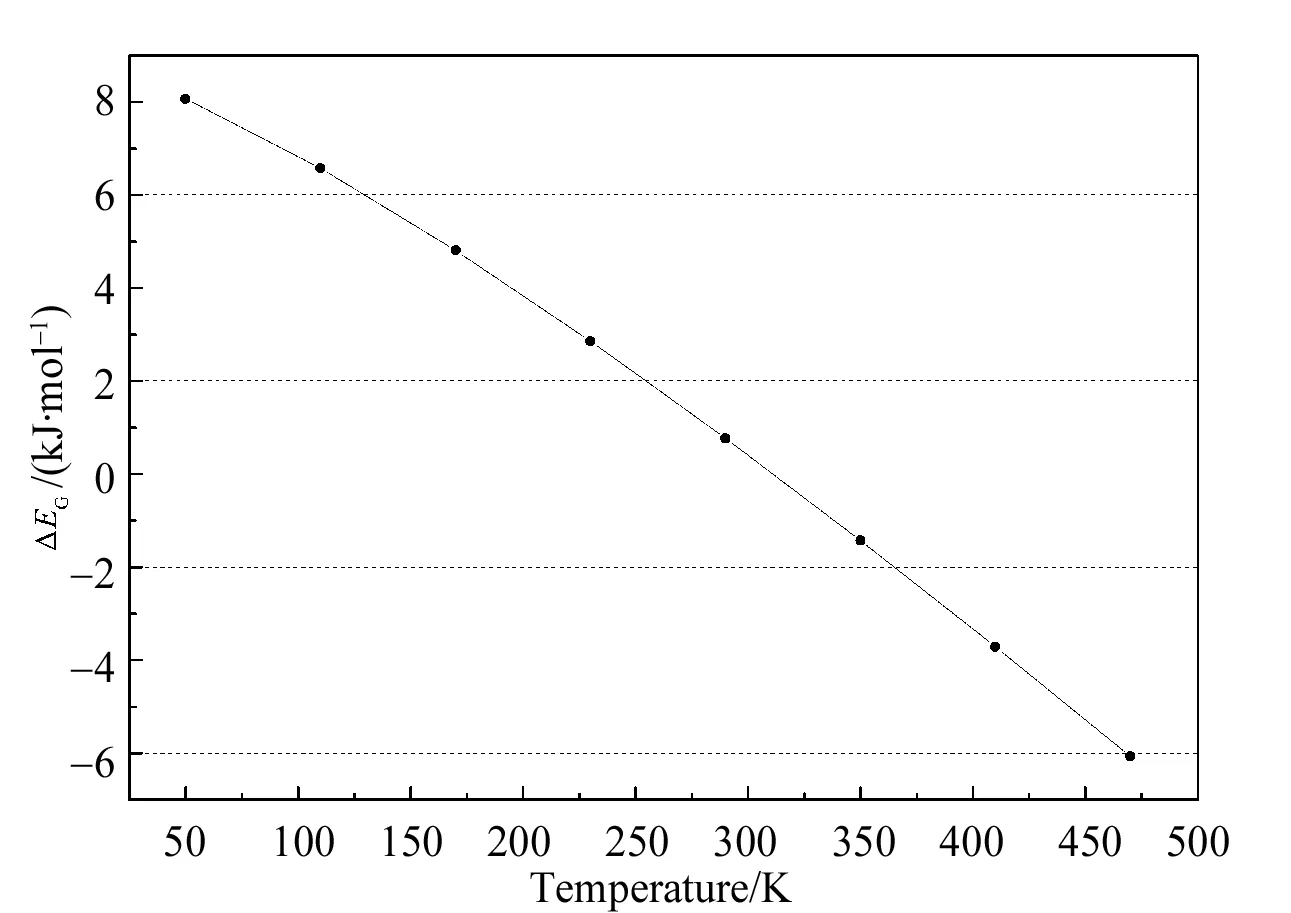
图4 Na4Si4·24H2的吉布斯相关的 平均吸附能随温度的变化Fig.4 Temperature dependence of averaged H2 adsorption energy with Gibbs free energy correction ΔEG for Na4Si4·24H2 complexes.
吉布斯自由能相关吸附能的温度依赖关系在一定程度上可反映出有利于H2吸附的温度范围。表3和图4可见,在低温至室温附近时,吉布斯相关吸附能量为正值;在高温时,吉布斯相关吸附能量为负值,表明低温有利于复合物的形成储氢,高温有利于复合物的解离释氢。这表明Na修饰Si4团簇对H2的吸附在较宽的温度范围内具有良好的热力学稳定性,即:在室温附近,从能量的角度有利于Na4Si4团簇对H2的吸附。因此,Na4Si4在室温及近室温可作为一类理想的储氢材料。
4 结 论
采用明尼苏达密度泛函M06方法研究了Na原子面位修饰正四面体Si4团簇的稳定结构及其与氢分子的相互作用, 结果表明:
1) Na原子吸附在Si4团簇表面不发生聚合, 前线轨道能隙表明面位修饰的Na4Si4体系具有较高的动力学稳定性。Na原子的修饰使H2与Na4Si4体系之间通过静电—偶极相互作用以及离子相互作用而吸附在钠原子周围, 改善了Si4团簇的吸氢能力。
2) 氢分子的平均吸附能范围为3.926~9.927 kJ·mol-1, 介于物理吸附与化学吸附之间; 理论计算的Na4Si4·24H2的储氢质量分数可达19.15%, 平均吸附能为9.927 kJ·mol-1,Na4Si4体系吸氢前后前线轨道能隙基本保持稳定,表明吸氢后的动力学稳定性基本不变,有利于体系可逆吸放氢过程的进行。
3) Na原子面位修饰正四面体Si4团簇具有较高的储氢密度和适宜的氢吸附能以及吉布斯相关吸附能。这表明:从理论上来说,Na4Si4在常温常压条件下可作为储氢材料。因此,本文为储氢材料的研制提供了研究思路,期待不久可以通过试验得到测试和验证。
猜你喜欢
中国特种设备安全(2022年4期)2022-07-08 02:41:40
中国特种设备安全(2022年4期)2022-07-08 02:41:28
物理学报(2021年12期)2021-07-01 09:42:56
数学物理学报(2020年6期)2021-01-14 01:00:46
青苹果·教育研究版(2016年6期)2016-12-02 09:15:30
武汉工程大学学报(2016年1期)2016-04-07 02:01:11
信息记录材料(2016年4期)2016-03-11 15:22:31
材料科学与工程学报(2016年5期)2016-02-27 07:11:37
华南师范大学学报(自然科学版)(2013年1期)2013-10-27 10:51:51
中学数学研究(2008年9期)2008-12-09 03:32:30
