
FISH技术在PML/RARA融合基因检测中的应用体会
2020-05-22 02:16罗秋萍
临床与实验病理学杂志 2020年2期
罗秋萍
急性早幼粒细胞白血病(acute promyelocytic leukemia, APL)是急性髓细胞白血病(acute myeloid leukemia, AML)的一种特殊类型(M3),其临床特点是出血倾向严重,常易并发弥散性血管内凝血及原发性纤溶亢进,早期病死率高,所以APL的早期诊断和及早治疗至关重要。现已明确t(15;17)是APL的特征性染色体异常,并在分子水平上导致PML/RARA融合基因的形成[1]。对APL患者进行PML/RARA融合基因的检测具有重要的临床价值,可为患者的诊断、疗效和预后提供有效帮助。采用荧光原位杂交(fluorescence in situ hybridization, FISH)技术对PML/RARA融合基因进行检测,具有成功率高、准确、省时,对检测标本限制低等优点[2]。因此,如何确保FISH的制片质量显得尤为重要。本科室经实践总结经验,对一些操作细节进行了改进,效果显著,现介绍如下。
1 材料与方法
1.1 材料 收集广州医科大学附属第三医院病理科2017年1月~2018年10月诊断的64例骨髓或外周血标本。
1.2 主要试剂 氯化钾、甲醇、冰乙酸、胃蛋白酶、HCl、无水乙醇、2×SSC溶液、NP-40溶液,探针选用广州安必平医药公司的PML/RARA融合基因检测试剂盒(粤食药监械生产许20111993号)。
1.3 主要仪器 徕卡显微系统(DM4B),恒温水浴箱,离心机,恒温烤箱,美国雅培StatSpin ThermoBrite自动原位杂交仪。
1.4 染色方法 (1)取骨髓或外周血2~3 mL(肝素钠抗凝)1 000 r/min离心12 min,用巴氏试管吸取中间的白膜层;(2)将吸取的白膜层细胞加入37 ℃水浴箱中提前预热的低渗液中(0.075 mol/L,5 mL),吹打混匀,于37 ℃水浴箱中低渗8 min;(3)往细胞低渗液中加入2 mL固定液(甲醇 ∶冰乙酸为3 ∶1),吹打混匀后离心(1 000 r/min,12 min);(4)去上清,加入5 mL固定液,吹打混匀后离心;(5)重复以上洗涤步骤,直至细胞沉淀呈白色,加入新鲜固定液,于4 ℃冰箱固定过夜;(6)第2天离心后去上清,根据细胞沉淀量加入合适新鲜固定液,吹打混匀后静置1 min,取10 μL细胞悬液分别滴加到3张干净载玻片上;(7)58 ℃恒温烤箱中烤片120 min以上;(8)在相差显微镜下选取细胞密度合适的玻片,并在玻片合适位置划出杂交区域,以方便后续操作;(9)将玻片放入胃蛋白酶工作液中消化4 min[工作液配制:50 mL纯化水中加入500 μL的HCl(1 mol/L),置于37 ℃恒温水浴箱中预热,使用前加入3 μL 胃蛋白酶浓缩液(20 mg/mL)];(10)取出玻片,将其放入10%中性福尔马林/PBS固定液中,室温固定4 min;(11)取出玻片置于2×SSC溶液中洗涤5 min;(12)取出玻片置入70%、80%、100%梯度乙醇中脱水各2 min,取出玻片,室温自然干燥;(13)避光环境中,加探针后放入自动原位杂交仪中进行杂交(78 ℃变性2 min,37 ℃杂交10~18 h);(14)第2天移去盖玻片,置于48 ℃预热的2×SSC溶液中洗涤4 min;(15)取出玻片置于48 ℃预热的NP-40溶液中洗涤4 min;(16)取出玻片置于70%乙醇,脱水3 min,取出玻片,室温自然干燥;(17)室温滴加DAPI复染液,使用荧光显微镜进行观察判读。
1.5 结果判读 荧光显微镜下GSP PML探针显示绿色信号,GSP RARA探针显示红色信号,若细胞显示为2红、2绿分散存在的荧光信号,则判定为阴性;典型阳性信号为1红、1绿、2黄,当出现2个以上或以下红色或绿色信号表示该基因拷贝数增多或丢失。
2 结果
本实验FISH法检测结果显示,64例标本中有57例标本获得高强度清晰的荧光信号(图1);5例标本因滴片细胞密度过高,细胞堆积在一起,细胞核上、核外杂信号较多,影响判读(图2);2例标本因未进行胃蛋白酶消化处理,整张玻片出现模糊和云雾状的背景(图3)。
3 讨论
FISH是一项在体外直接观察细胞中特定的核酸技术,原理是利用已知的标记单链核酸为探针,按照碱基互补原则,与待检材料中未知的单链核酸进行特异性结合,形成可被检测的杂交双链核酸[3]。FISH检测的关键是清晰地观察到荧光杂交信号,但在激发光照射下,标本背景成分(如核蛋白、胞质蛋白、胶原纤维等)均可产生非特异性背景荧光(未洗除的荧光素标记探针也包含在内),一旦背景荧光过强,则会影响目标荧光信号的判读。因此,FISH检测的关键除了保证杂交成功外,还需减弱背景荧光,增强目标荧光信号,即提高信噪比[4]。作者在实践中发现,细节操作的改进可以提高信噪比,主要有以下几点:(1)确保白细胞收集的质量:有文献报道白细胞的收集是直接离心去上清后加入0.075 mol/L低渗液,低渗后加入固定液反复清洗直至细胞沉淀呈白色[5]。虽然肉眼可见细胞沉淀呈白色状态,但由于带入血液标本中的红细胞,即使红细胞被裂解,但残留的细胞碎片会产生较多的非特异性背景荧光。肝素钠抗凝的骨髓或外周血标本经过离心,由于比重不同,会出现分层现象,上层淡黄色半透明液体为血浆,中间薄薄一层白色物质为白细胞和血小板,下层暗红色不透明的为红细胞。PML/RARA融合基因的检测只需要收集白细胞,所以实验中只吸取中间的白膜层即可,减少红细胞的带入,降低非特异性背景荧光。(2)细胞悬液制备过程的离心速度是关键:多数文献报道制备细胞悬液时离心速度一般为1 500~2 000 r/min[5-6]。本科室总结多年经验发现细胞核在未彻底固定好的情况下,进行高速离心,可能会发生损坏,直接导致荧光杂交信号分散不集中,甚至无法判读,且这种信号分散的情况是不可逆的[7]。所以细胞悬液制备过程中一般会将离心速度设定在1 000 r/min,离心时间相对延长(12 min)。(3)制片质量:取1张干净的载玻片,重悬细胞后取合适悬液滴加到载玻片上,室温晾干后用10×物镜在相差显微镜下观察细胞密度,要求细胞无重叠,且单视野细胞数量在100~200个为宜,如细胞密度不合适,则根据要求重新制片。若细胞重叠较多,影响信号判读,同时容易影响消化效果产生较多信号杂点;若细胞密度过低,计数达不到100个细胞,同样会影响判读。(4)胃蛋白酶消化时间的把握:玻片在进行杂交前一般需要进行胃蛋白酶的消化处理,消化的目的是通过微小组织蛋白降解,可增加细胞的渗透性从而增加探针的穿透力。如果消化不足会使核酸无法充分暴露,而消化过度则会破坏核酸周围的蛋白结构,导致后续步骤中核酸的丢失[8]。骨髓或外周血标本与石蜡组织切片标本的最大区别:前者细胞分散存在,细胞之间没有过多交联。消化处理的酶一般选用胃蛋白酶,并且浓度相对较低,消化时间可按预试验进行调整。一般消化完成的细胞可在未加探针之前,使用荧光显微镜DAPI通道进行观察,以便及时了解消化效果。消化程度适当的细胞核亮度合适、核膜完整、边缘光滑,通过微调感觉到细胞有立体感,核表面有明显的凹凸不平感,视野清晰;如果细胞核亮度灰暗,产生黑洞和孔状,核膜呈锯齿状一般视为消化过度;如果细胞核亮度刺眼,通过微调,细胞立体感不强,细胞视野比较模糊、轮廓不清、表面有云雾状一般视为消化不足[9]。(5)后固定的必要性:细胞片经胃蛋白酶消化后,采用10%中性福尔马林/PBS固定液进行后固定,固定时间4~5 min。后固定的主要目的是防止核酸在消化处理后,在后续的实验步骤中丢失,同时可以使杂交信号更加集中清晰,利于观察判读。如果后固定液的浓度过高或固定时间过长,则有可能会使蛋白重新发生交联,令杂交信号背景升高,不利于信号观察判读[7]。
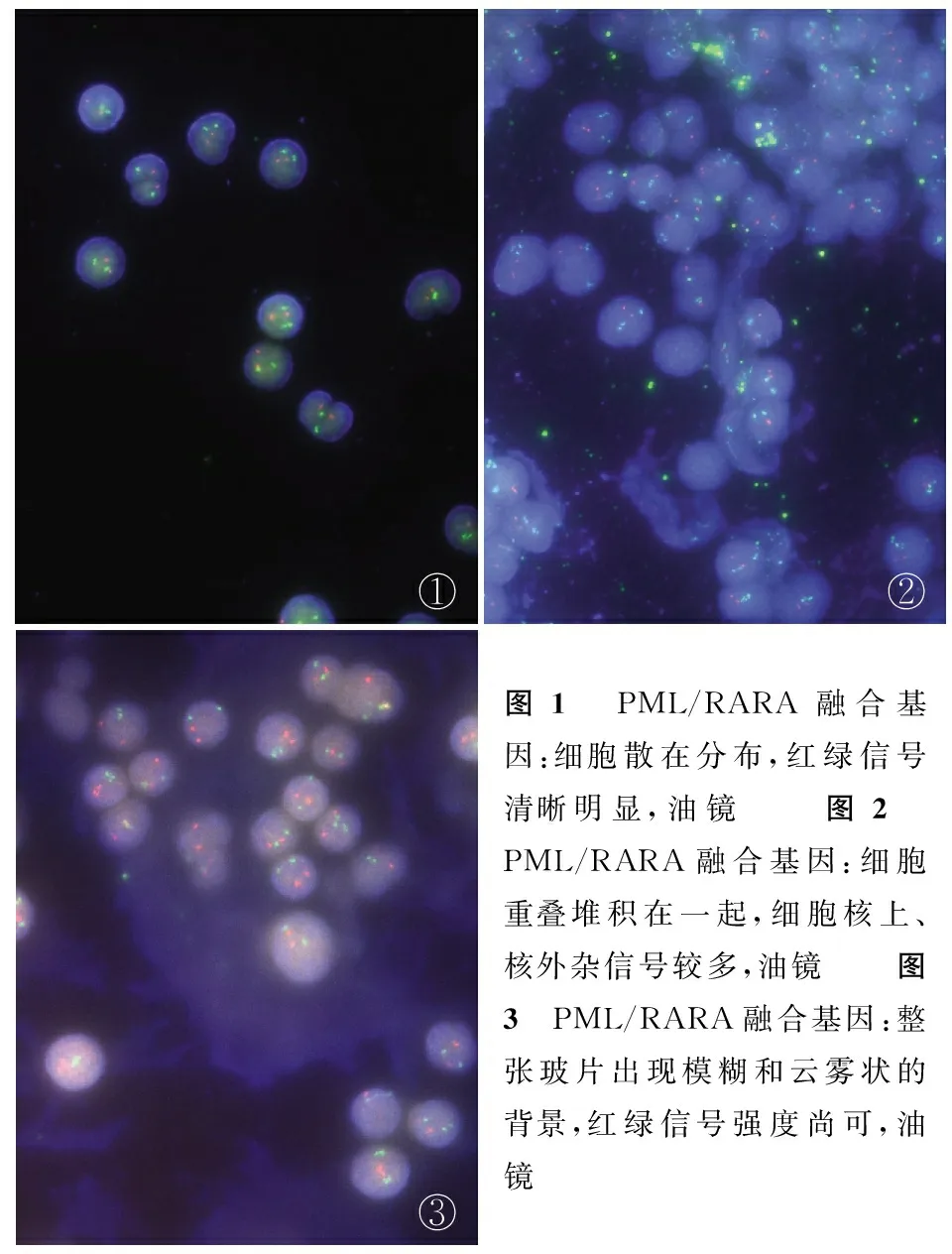
①②③图1 PML/RARA融合基因:细胞散在分布,红绿信号清晰明显,油镜 图2 PML/RARA融合基因:细胞重叠堆积在一起,细胞核上、核外杂信号较多,油镜 图3 PML/RARA融合基因:整张玻片出现模糊和云雾状的背景,红绿信号强度尚可,油镜
综上所述,在FISH实验中影响荧光信号的因素较多,要制出一张高质量的FISH切片,需要准确把握每一步骤的实验条件,并谨慎操作。
猜你喜欢
中国慈善家(2022年3期)2022-06-14
现代苏州(2022年9期)2022-05-26
快乐语文(2021年34期)2022-01-18
装备制造技术(2020年1期)2020-12-25
中国(俄文)(2020年8期)2020-11-23
临床检验杂志(2020年1期)2020-03-02
科技与创新(2019年5期)2019-03-22
分析化学(2018年4期)2018-11-02
现代检验医学杂志(2016年3期)2016-11-15
现代检验医学杂志(2014年5期)2014-02-02
