
V-Ce 复合氧化物催化间二甲苯气相氨氧化制间苯二甲腈
2020-05-22 02:24习鹏博顾龙勤杨为民
化学反应工程与工艺 2020年3期
习鹏博,冯 冰,顾龙勤,杨为民
1. 华东理工大学化工学院,上海 200237;2. 中国石油化工股份有限公司上海石油化工研究院 绿色化工与工业催化国家重点实验室,上海 201208
间苯二甲腈(isophthalonitrile,简称IPN)是合成塑料、纤维、农药(百菌清)以及环氧树脂固化剂等产品的重要原料,可经多种方法制备。其中最简单经济的方法是以间二甲苯、空气和氨气为原料,在催化剂的作用下,经过气相氨氧化反应一步合成间苯二甲腈。用于芳烃气相氨氧化反应的催化剂主要有V 系和Sb 系,其中V 元素氧化物具有较为理想的晶格氧性能,因而得到了广泛应用[1]。此外,Cr 是常用的V 系催化剂改性元素,V-Cr 体系催化剂具有制备简单、重复性好以及芳烃产品收率高等优点,因此在工业流化床反应器上应用较多[2-3],但Cr 是一种具有致癌作用的重金属元素,随着绿色化工的发展,开发无Cr 的V 系催化剂具有重要意义。
Ce 作为稀土元素中储量最大的金属,具有无毒且相对便宜等优点,其氧化物对具有较强的储氧以及加快氧化还原循环能力[4]。考虑到间二甲苯氨氧化是一个连续脱氢、供氧并腈化[5]的过程,在V系催化剂中添加Ce 可以调变催化剂晶格氧的活性,促进反应的进行,掺杂少量的Mo 元素还可以改变V=O 键能,对加快产物从催化剂表面脱附,减少过度氧化有促进作用[5]。
因此,本工作在固定催化剂主载比的条件下(活性组分与载体质量比为1:1),通过改变活性组分中Ce 和V 的物质的量之比(Ce/V 比)来合成不同的V-Ce 复合氧化物催化剂,考察其氨氧化催化性能,寻求高性能催化剂的最佳Ce/V 比。在此基础上,添加助剂Mo 元素,进一步提高间苯二甲腈收率。
1 实验部分
1.1 催化剂制备
将一定量五氧化二钒称重后加入草酸水溶液中,加热搅拌1 h 得到蓝色混合溶液A;随后将硝酸铈及助催化剂分别溶于水中,并加入到A 溶液中,之后与载体硅溶胶进行充分混合,80 ℃下加热搅拌形成稳定的前驱体混合液,再经喷雾成型,500 ℃下焙烧10 h 得到催化剂V1CexMoy/SiO2,其中x和y 分别为相应元素与V 的物质的量之比(x 分别为1.5,1.0,0.8 和0.6;y 分别为0.05,0.10 和0.15)。
1.2 催化剂表征
采用Bruker D8 Avance A25 型X 射线衍射仪(XRD)对催化剂的晶体结构进行分析,Cu Kα 辐射(λ 为0.154 06 nm),电流200 mA,管电压40 kV,扫描范围5~80º,扫描速率12 (º)/min。采用Jobin Yvon LabRam-1B 型拉曼光谱仪对催化剂进行拉曼光谱(Raman)分析,光源为He-Ne 激光器(632.8 nm),功率为64 mW,CCD 检测器。采用Micrometritics Auto Chem ΙΙ 2950 型程序升温化学吸附仪对催化剂进行氢气程序升温还原(H2-TPR)分析,称取0.1 g 催化剂样品置于U 型石英管中,先在200 ℃高纯氩气下吹扫1 h,然后降至室温,在H2/N2氛围下,以10 ℃/min 的升温速率升至900 ℃。采用Micrometritics Auto Chem ΙΙ 2920 型程序升温化学吸附仪对催化剂进行氨气程序升温脱附(NH3-TPD)分析,催化剂装填量为0.15 g,将样品在550 ℃下采用He 吹扫30 min,降温至60 ℃左右,通入NH3(20 mL/min)和He(30 mL/min)30 min,然后关闭NH3阀,升温至100 ℃,用He 吹扫40 min 以除去物理吸附的NH3,然后以10 ℃/min 的升温速率升至600 ℃,用热导检测器分析脱附的NH3。采用Perkin Elmer phi 5000c 型X 射线光电子能谱仪(XPS)对催化剂表面的元素组成及状态进行分析,Mg Kα 射线源(1 253.6 eV),工作电压15 kV,工作电流20 mA,所有能谱以C1s 为284.8 eV 为标准进行校准。
1.3 催化剂评价
间二甲苯(mX)气相氨氧化反应在湍流流化床反应器中进行,反应温度为425 ℃,反应压力为120 kPa,进料mX,NH3和空气物质的量之比为1:7:35,催化剂质量空速(WHSV)为0.067 h-1。待通入原料并控制到达反应温度后,稳定2 h,随后进行取样,取样时间同样为2 h。IPN 产品采用薄壁冷凝器收集,反应后的所有产物以气态的形式首先进入薄壁冷凝器,其中IPN 等有机相产物在冷凝器中降温、凝结成固体,该部分有机相产物通过水洗、抽滤和离心后称重得到含水样品质量;随后取10 g 含水样品,加入到盛有100 mL 甲苯溶液的圆底烧瓶中,烧瓶出口分别连接分水器和冷凝管,放置于电热套上加热回流,测定其含水量;接着取少量固体产物溶于丙酮中,以气相色谱分析其中不同有机相的比例,根据分析的结果综合计算不同产物的单程收率。
在反应过程中产生的尾气经盛有硫酸溶液的吸收瓶洗涤后,以气袋收集气相中的不凝部分,并通过Agilent 7890A 型气相色谱对COx等气相产物进行离线分析,3 m×Ø2 mm 分子筛填充柱,柱温180 ℃,TCD 检测器。
2 结果与讨论
2.1 催化剂的表征结果
不同Ce/V 比催化剂的XRD 图谱如图1 所示。由图可知,催化剂在2θ 为18.1,24.0,30.3,32.4,34.3,39.0,43.5,46.4,47.9,49.2,55.5,56.5,60.2,62.4,67.9 和71.1º 处,均出现明显的衍射峰。经对比,与CeVO4标准卡片(JCPDS 12-0757)相对应,表明4 个催化剂中均含有明显的CeVO4晶相。此外,V1Ce1和V1Ce1.5在2θ 为28.5,33.1,47.5 和56.3º处出现不同强度的衍射峰,对比标准卡片(JCPDS 43-1002),发现这些衍射峰归属于CeO2晶相。进一步比较4 个不同Ce/V 比催化剂的XRD 图谱,可以发现V1Ce0.8催化剂的CeVO4晶相的衍射峰最强,证明其CeVO4晶相含量最高,继续增加催化剂中Ce 的含量,CeO2晶相出现且衍射峰强度明显增加。

图1 不同Ce/V 的V-Ce 催化剂的XRD 图谱Fig.1 XRD spectra of V-Ce catalysts with different Ce/V ratios
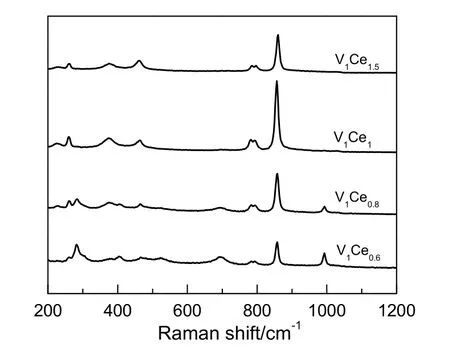
图2 不同Ce/V 的V-Ce 催化剂的Raman 图谱Fig.2 Raman spectra of V-Ce catalysts with different Ce/V ratios
通过对不同Ce/V 比催化剂进行拉曼光谱表征分析,进一步确认了催化剂中各物种的结构信息,结果如图2 所示。由图可知,V1Ce0.6和V1Ce0.8催化剂在990 cm-1处均存在对应于V2O5物种中V=O的拉曼谱峰,而在XRD 图谱中基本未发现V2O5,说明在低Ce 含量时仍存在一部分V 氧化物物种,但由于这部分V 物种分布较为均匀,因此未出现明显的XRD 衍射峰。所有催化剂在260,369,460,780 和855 cm-1处均出现一定强度的拉曼谱峰,这部分谱峰对应的是CeVO4物种,其中855 cm-1处的拉曼谱峰对应于VO43-物种中的A1g对称伸缩振动(ν1);780 和793 cm-1处的拉曼谱峰对应于VO43-物种中的B1g和Eg反对称伸缩振动(ν3);460,369 和260 cm-1对应于B1g(ν4),A1g(ν3)与B1g(ν3)形变,证明上述催化剂中均存在CeVO4相。此外,460 和780 cm-1的拉曼谱峰也可归属于CeO2物相[6-8],从图2 中还可以发现,随着Ce 含量的提高,这两处的拉曼谱峰强度有着很明显的增长趋势,与XRD表征结果相符。
不同Ce/V 比催化剂的H2-TPR 表征结果如图3所示。由图可知,所有催化剂均具有两个主要的H2还原峰,其中530 ℃左右的还原峰面积较小,对应于载体作用力较弱,容易还原的表面V5+和Ce4+物种,这类物种含量较低,而较高温度的还原峰在650~800 ℃,对应的是催化剂体相物种的还原,尤其是以CeVO4到CeVO3的贡献为主[9],同时也包括体相CeO2等物种的还原,由于这部分体相物种与载体作用力较强,因此其还原峰温度较高。通过对比不同Ce/V 比催化剂的H2-TPR 图谱可以看到,随着Ce 含量的增加,V-Ce 催化剂在530 ℃的还原峰出峰温度逐渐降低,这是由于表面Ce 物种的增多削弱了SiO2载体与V 和Ce 氧化物之间的相互作用而引起的,这部分物种变得易于还原后,对氧化反应的循环是存在促进作用的。但是同时在高温还原峰的区域内,V1Ce0.6原本具有类似于两个肩峰合并而成的还原峰,对应体相CeVO4中V5+和Ce4+的还原[10],随着Ce 含量的增加,680 ℃的峰在Ce/V 比为0.8 时仍保持位置不变,但另一处肩峰还原温度进一步升高,到Ce/V 比为1.0 和1.5 时,两个肩峰均变得更为平坦,且进一步向高温移动,说明体相CeO2的晶体尺寸持续增加,还原难度进一步增大,这对于氧化反应活性位的再生循环是不利的。综合来说,CeO2晶相的生成促进了表面物种的还原,但其存在于体相本身,使得更多部分的体相CeVO4还原难度加大,影响了催化剂性能。

图3 不同Ce/V 的V-Ce 催化剂的H2-TPR 图谱Fig.3 H2-TPR patterns of V-Ce catalysts with different Ce/V ratios
图4 为不同Ce/V 比催化剂的NH3-TPD 图谱。从图中可以看出,催化剂的酸性位主要归属于弱酸,其脱附温度大约为175 ℃左右,并且该处的脱附温度基本没有随着Ce 含量的改变呈现改变的趋势。其中V1Ce0.6和V1Ce0.8两个催化剂峰型对称性相对较好,无明显拖尾;而进一步增大Ce 含量后发现,V1Ce1和V1Ce1.5两个催化剂在200 ℃以后出现了较小的峰突起,说明随着Ce 含量增加,酸强度有所提高。考虑到芳烃气相氨氧化反应中,催化剂酸性过强将导致深度氧化,而酸性过弱则不能为反应中被活化的甲基提供N 物种,因此酸性的增强不利于气相氨氧化反应主产物IPN 选择性的提高。
通过对催化剂进行XPS 表征,研究其表面活性组分的分布状态,结果如图5,6 和7 所示。

图4 不同Ce/V 的V-Ce 催化剂的NH3-TPD 图谱Fig.4 NH3-TPD curves of V-Ce catalysts with different Ce/V ratios

图5 不同Ce/V 比的V-Ce 催化剂Ce 3d 的XPS 图谱Fig.5 XPS spectra of Ce 3d in the V-Ce catalysts with different Ce/V ratios
Ce 的XPS 图谱可以分峰为Ce 3d3/2和Ce 3d5/2,其中v0,v',u0和u'归属于Ce3+的特征峰,v,v',v''',u,u''和u'''归属于Ce4+的特征峰[11-13],在Ce/V比为0.6,0.8 和1.0 时,Ce4+的特征峰中只有v,v''',u,u'''这两组峰,当Ce/V 比为1.5 时,Ce4+出现v'',u''这组峰,结合表1 中的数据分析得知,催化剂表面的Ce4+/Ce3+比随着催化剂中Ce 含量的增加而逐渐增大,当Ce/V 比为0.6,0.8 和1.0 时,催化剂表面的Ce4+均少于Ce3+,继续增加活性组分中Ce/V 比到1.5,催化剂表面Ce4+/Ce3+比增大为1.02,这同时反映在了催化剂中CeO2含量的增高,与XRD 表征结果相符,由此可见,v''和u''这组峰与催化剂表面的Ce4+含量有一定的关系。催化剂表面的V 主要以V5+的形式存在,V 2p3/2结合能在517~518 eV[14],随着Ce/V 比的增加,V 2p3/2结合能呈现先减小后增加的趋势,当Ce/V 比为0.8 时,V 2p3/2结合能最小为517.4 eV,V 作为反应的活化中心,较低的结合能更有利于底物的活化。催化剂表面O 的状态如图7 所示,O 1s 在530.7 eV 的峰来自于晶格氧(Ce–O 和V–O),533 eV 的峰来自于催化剂表面的吸附氧(Si–OH),由于使用SiO2做载体,所以催化剂表面吸附态氧的含量远大于晶格氧,相比于晶格氧,吸附态氧的氧化活性更高,这是由于其具有更高的迁移率、更多的表面氧空位和缺陷位点[15-16]。
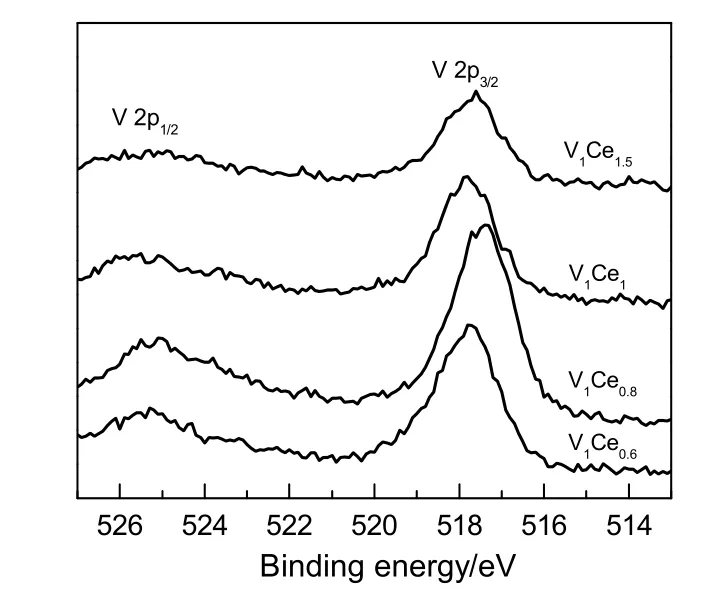
图6 不同Ce/V 比的V-Ce 催化剂V 2p 的XPS 图谱Fig.6 XPS spectra of V 2p in the V-Ce catalysts with different Ce/V ratios

图7 不同Ce/V 比的V-Ce 催化剂O 1s 的XPS 图谱Fig.7 XPS spectra of O 1s in the V-Ce catalysts with different Ce/V ratios
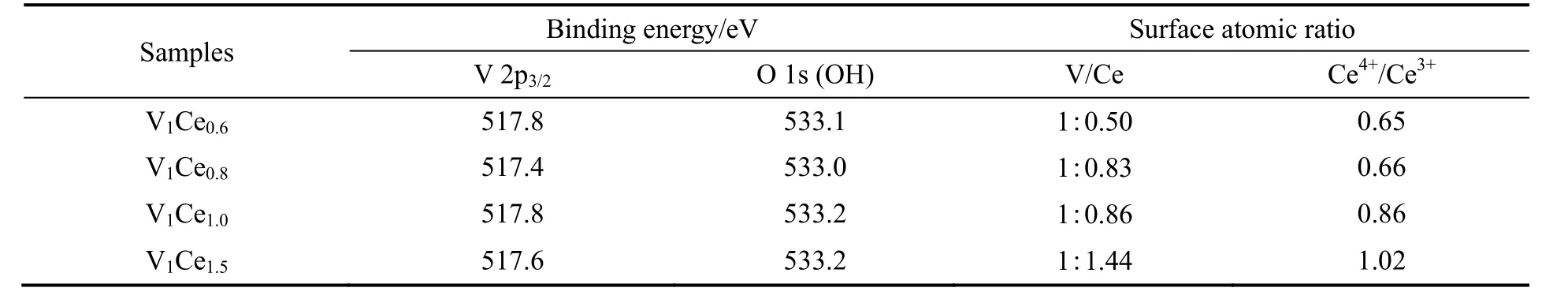
表1 不同Ce/V 比的V-Ce 催化剂的XPS 分析Table 1 XPS analysis of V-Ce catalysts with different Ce/V ratios
2.2 不同Ce/V 组成催化剂的气相氨氧化反应性能
在流化床反应器中对上述催化剂进行气相氨氧化反应测试,表2 列出了其催化间二甲苯氨氧化的反应性能数据。分析反应产物发现,有机相产物主要以IPN 和间二甲苯不完全氨氧化的副产物间甲基苯甲腈(MTN)为主,而气相产物中主要为深度氧化反应产生的CO2和CO(COx)。预期中的苯甲腈与氢氰酸等副产物的含量较低,因此表中主要列举了间二甲苯的转化率,间甲基苯甲腈、间苯二甲腈和COx的选择性,并以此作为催化剂反应性能评价的比较标准。
从表2 中可以发现,所有考察范围内的不同Ce/V 比催化剂,其间二甲苯转化率接近100%,在有机相产物收集过程中未发现存在间二甲苯。主要有两点原因:一是反应的进料mX,NH3和空气物质的量之比为1:7:35,相较于间二甲苯,氨气和空气都是过量的,有利于间二甲苯的完全转化;二是V元素的氧化物具有充分的晶格氧,尤其是在添加Ce 元素后,作为一种具有吸氧性能的稀土元素,复合氧化物催化剂的氧化能力有进一步的提高,因此该体系催化剂具备较强的有机分子反应物活化性能。从产物分布来看,随着催化剂中Ce/V 比例的提高,主产物IPN 的选择性呈现先增大后减小的趋势,当Ce/V 比为0.8 时,IPN 选择性达到最高,单程收率达到了60.0%,继续增加催化剂的Ce/V 比,IPN收率迅速下降至26.8%,而同时COx的选择性由34.1%增加至41.8%,证明随着Ce 含量的进一步增加,催化剂的氧化活性逐步提高,而当其氧化活性过高时,会对主产物IPN 选择性造成不利影响,因此存在最优的Ce/V 比为0.8。
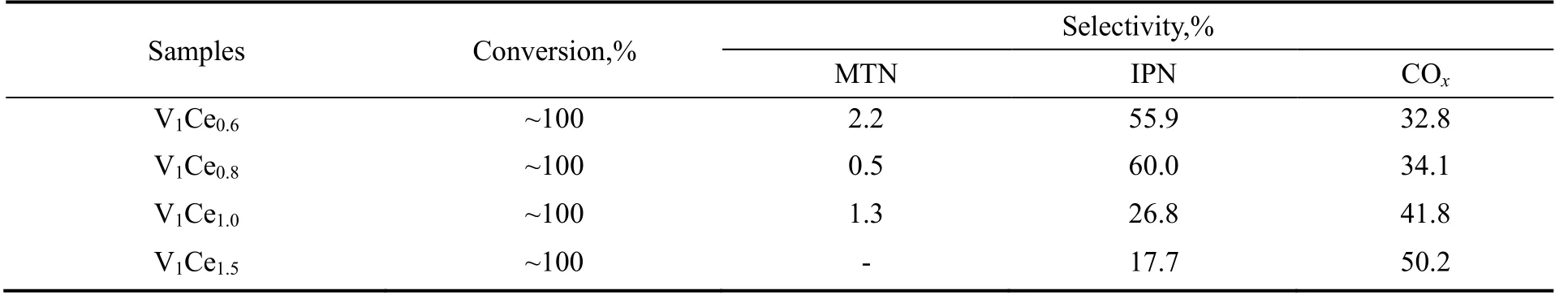
表2 不同Ce/V 比对V-Ce 催化剂间二甲苯气相氨氧化制备间苯二甲腈反应性能的影响Table 2 Effect of different Ce/V ratios on the catalytic performance of V-Ce mixed oxide in the ammoxidation of m-xylene
2.3 助剂Mo 对V-Ce 复合氧化物催化剂性能的影响
在筛选出最佳的Ce/V 比的基础上,考虑到Mo 元素的添加可以调节V 系氧化物催化剂中V=O键强度,提高催化剂选择性,因此选择将Mo 元素作为助剂添加到V1Ce0.8催化剂体系中,采用Mo/V比为0.05,0.10 和0.15 进行考察。表3 中列出了在V1Ce0.8催化剂基础上,不同Mo 添加量的V-Ce系催化剂上间二甲苯氨氧化反应性能评价结果。结果显示,所有考察范围内催化剂的间二甲苯转化率近100%,证明添加Mo 后的V-Ce 催化剂仍具有较高的间二甲苯氧化活性。从产物分布来看,IPN 收率随着Mo 添加量的提高有一定的提升,尤其是在Mo/V 比为0.10 时,收率达到最高的62.5%,说明Mo 的添加更主要的是从提高反应选择性来对催化剂进行改善。从副产物上看,添加助剂Mo 的催化剂COx收率均低于未添加Mo 的V1Ce0.8催化剂,说明Mo 的添加从反应物分子活化的角度抑制了一部分间二甲苯的深度氧化,从而提高了催化剂的IPN 选择性。

表 3 Mo 添加量对V1Ce0.8 催化剂间二甲苯气相氨氧化制间苯二甲腈反应性能的影响Table 3 Effect of Mo addition on the catalytic performance of V1Ce0.8 composite oxide in the ammoxidation of m-xylene
3 结 论
以SiO2为载体,喷雾成型法制备的V-Ce 复合氧化物催化剂对间二甲苯氨氧化反应具有良好的催化活性,通过表征发现,催化剂的主要活性相为CeVO4,当Ce/V 比为0.6 和0.8 时,有少量分布均匀的V2O5存在,Ce/V 比为1.0 和1.5 时,出现具有强氧化活性的CeO2晶相。催化剂表面呈弱酸性,V 在催化剂表面主要以V5+形式存在,而Ce 以Ce4+和Ce3+形式共存,同时有大量的吸附态氧(OH)存在。在温度为425 ℃,压力为120 kPa,催化剂质量空速为0.067 h-1,进料mX,NH3和空气物质的量之比为1:7:35 的反应条件下进行评价,在V-Ce 催化剂中,V1Ce0.8性能最优,IPN 收率为60.0%。添加助剂Mo元素,可显著抑制氧化现象,降低COx选择性,并在V,Ce和Mo物质的量之为1.0:0.8:0.10时,IPN 收率达到最大值62.5%。
猜你喜欢
世界农药(2022年10期)2022-11-10
炼油技术与工程(2022年8期)2022-08-18
供水技术(2022年1期)2022-04-19
能源化工(2021年2期)2021-12-30
陶瓷学报(2021年4期)2021-10-14
粉末冶金技术(2021年3期)2021-07-28
农药科学与管理(2021年2期)2021-03-16
保鲜与加工(2021年1期)2021-02-06
英语文摘(2020年7期)2020-09-21
今日财富(2017年32期)2017-10-19
