
CIB2基因在遗传性聋中的研究进展*
2020-05-20 03:38陈梦兰李云龙
听力学及言语疾病杂志 2020年2期
陈梦兰 李云龙
现已发现约300多个基因与听力发生过程相关,其中,钙整合素结合蛋白2(calcium- and integrin-binding protein 2,CIB2)基因最近在遗传性耳聋相关致病基因研究中被频繁提及,目前发现的大多数CIB2基因突变确定与耳聋相关,主要导致非综合征型聋DFNB48和综合征型聋USH1、USH1J综合征[1~6]。此外,CIB2基因还可能与发育迟缓、肥胖、卵巢癌、艾滋病、智力障碍、心血管系统异常等疾病相关。CIB2基因结构、功能、动物模型及致病机制等研究对遗传性聋的产前诊断与筛查、早期预防和治疗等具有重要意义,然而CIB2基因的许多功能及致病机制等仍未完全阐明,故本文对CIB2基因在遗传性聋中的研究现状进行综述,以期能为临床和科研工作者提供参考。
1 CIB2基因概述
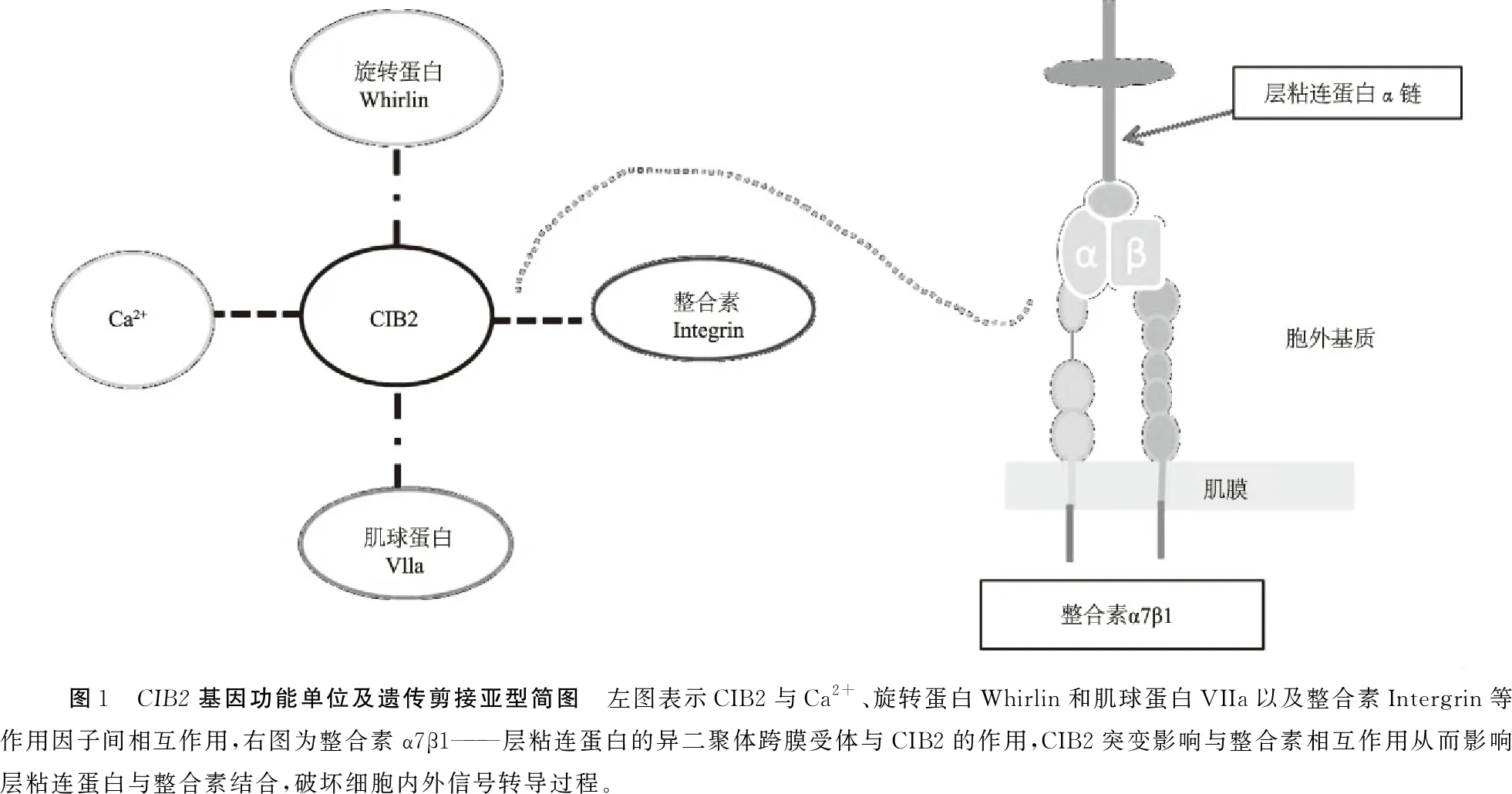
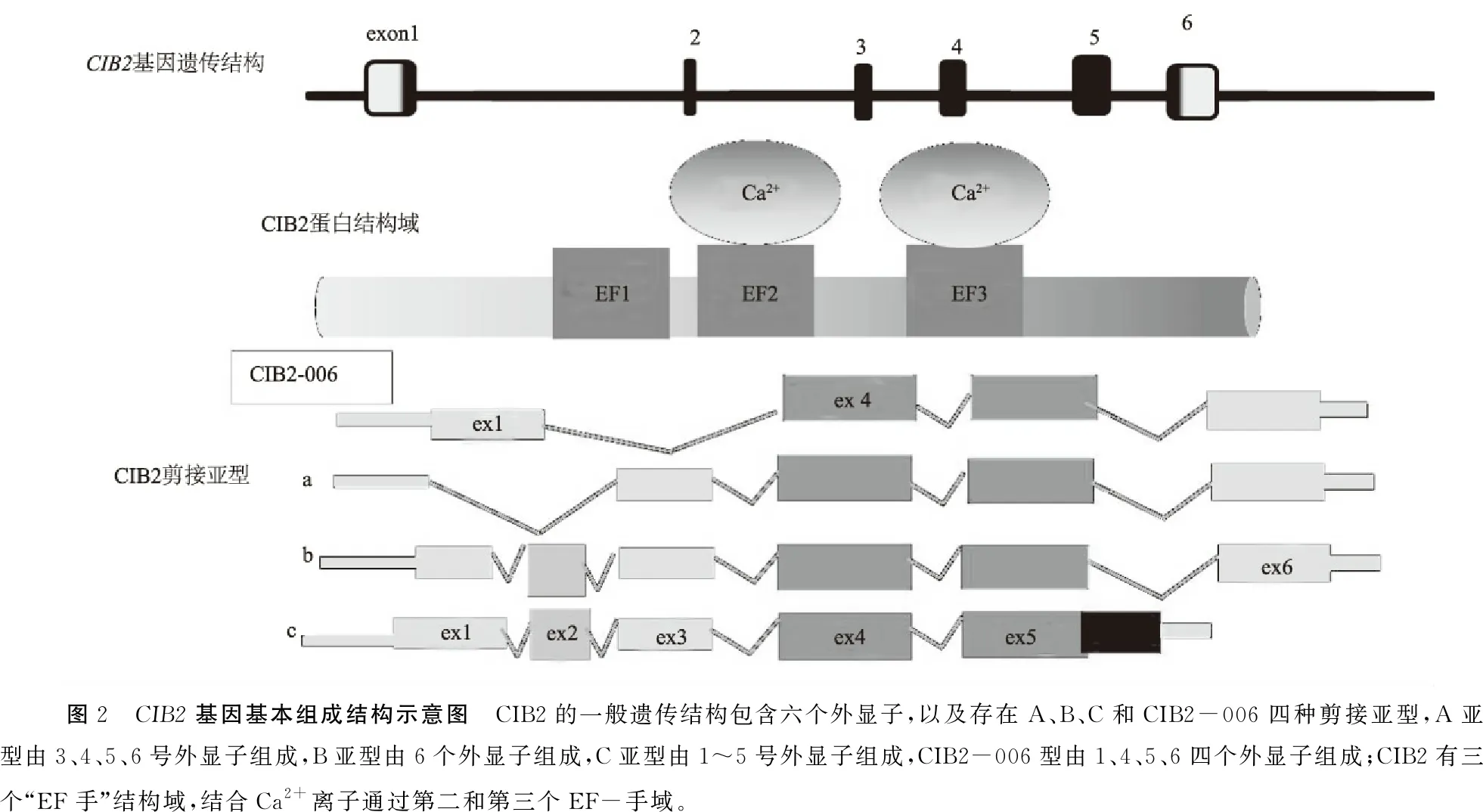
1.1CIB2基因的结构及功能 对CIB基因家族的研究始于20世纪90年代,当时有两个独立研究小组报道了一种根据其特定功能命名的新蛋白——钙整合素结合蛋白(calcium- and integrin- binding proteins,CIB)[7]或激酶作用蛋白(kip)[8]。CIB/Kip蛋白与钙调蛋白和钙调神经磷酸酶B具有同源序列,与核DNA依赖性丝氨酸/苏氨酸蛋白激酶相互作用,且呈现钙合整合素结合活性[7~10]。现已发现CIB家族四个成员,依据其发现先后顺序分别命名为CIB1、CIB2、CIB3 和CIB4[11,12],它们都具有一个特殊功能单位——“EF手”[11]。“EF手”是一种可以与Ca2+结合的“螺旋-环-螺旋”变体,由11~14个氨基酸残基组成。Ca2+与CIB蛋白结合是通过两个“EF手”之间的β-支架固定Ca2+,然后“EF手”扭转自身结构以协调Ca2+与环氨基端和羧基端结合的平衡使其达到稳定三维构象。CIB2基因(OMIM:*605564)位于15号染色体,其定位约为15q25.1,具有四种由4~6个外显子组成的剪接异构型[2](图1)。CIB2含有三个“EF手”结构域,仅“EF2”和“EF3”能够结合Ca2+[11],且在与Ca2+结合时发生构象变化并产生疏水口袋,介导CIB2与整联蛋白尾部C末端相互作用,从而调节钙离子稳态(图1,2)。
1.2CIB2相互作用因子 CIB2通过“EF手”结构域结合Ca2+后,使CIB2蛋白构象转变为Ca2+结合形式,这是蛋白质传递Ca2+信号的特征之一,“EF手”型Ca2+结合蛋白在Ca2+信号转导过程中起着重要作用,如:控制Ca2+通道的功能、调节Ca2+信号强度和持续时间以及对突触前内毛细胞功能和内耳MET的调节作用[11,13~16]。CIB2与旋转蛋白(Whirlin; 607928)和肌球蛋白VIIa(MYO7A; 276903)相互作用并多聚化,将会导致USH综合征,但目前仍未发现关于这种相互作用之后的具体机制信息的报道。CIB2基因与整合素相互作用从而影响细胞内外信号转导及细胞生长发育的调控等过程,整合素是细胞粘附和激活细胞内、外信号通路的关键,它们是细胞外基质中配体的α/β异二聚体跨膜受体(图1),整合素功能障碍可能是导致许多疾病的原因[17]。整合素控制着立体纤毛成熟和毛细胞分化的过程,通过调节肌动蛋白动态决定F-actin细胞骨架,因此,整合素是听觉通路中必不可少的元件之一[18]。
1.3CIB2基因人体的定位表达研究 CIB2蛋白在人体多种组织和器官中均有分布,如骨骼肌、大脑、眼睛和内耳[2]。1999年,对人体组织的非定量逆转录PCR分析发现,CIB2 mRNA在脑、肺、骨骼肌和血小板[7]等多种人体组织中均有转录,但当时没有发现CIB2在人耳和视网膜中表达。骨骼肌、肝脏、脑、脾、心、肾、肺的半定量和实时定量PCR结果表明,CIB2 mRNA主要分布在骨骼肌的肌腱连接和神经肌肉接头处,相对较少分布在脑组织的海马区和肺中[19]。
2 CIB2基因突变
2.1CIB2基因突变CIB2基因突变首先在巴基斯坦和土耳其人群中被发现,后在荷兰、西班牙、伊朗等人群也有发现。迄今为止,已发现的CIB2基因突变约40多种,且所有突变都存在变异模式遗传,其中常见的CIB2基因耳聋相关突变主要有:c.97C>T(p.Arg33*),c.192G>C(p.Glu64Asp),c.196C>T(p.Arg66Trp),c.272T>C(p.Phe91Ser),c.297C>G (p.Cys99Trp),c.368T>C(p.Ile123Thr)和c.556C>T(p.Arg186Trp)等[2,5,6,20],多突变位点的致病性见表1和图3。目前中国常见的CIB2基因致聋突变主要是c.196C>T、c.272T>C、c.297C>G等,但国内耳聋患者CIB2基因突变型和突变携带率等报道较少。CIB2基因剪接亚型有四种,即A、B、C型和CIB2-006型(图2)。c.97C>T突变影响异构型B和C而不影响A和CIB2-006型[2,6],而c.556C>T可能影响异构型A、B和CIB2-006型[5],其他突变均不影响A、B、C三种异构型。2017年Michel等[21]报道了两个新的c.34C>T(p.Gln12*)和c.330T>A(p.Tyr110*)无义突变,患者表现出重度非综合征型聋,且未表现出前庭和视网膜功能障碍,c.330T>A突变定位于Exon4中,影响所有剪接亚型的编码序列,c.34C>T影响异构型CIB2-006、B和C型[22]。
c.272T>C、c.297C>G突变可破坏Ca2+效应结合位点,而c.368T>C突变可能放大了Ca2+结合亲和力。c.192G>C、c.272 T>C和c.297C>G突变可能降低CIB2与整合素的相互作用。c.556C>T突变影响CIB2的尖端定位及其与旋转蛋白的相互作用。c.34C>T突变导致蛋白质第12位密码子过早终止[22],而c.97C>T位点突变可能导致含有过早终止密码子的特异性mRNA降解[23]。Vallone等[24]研究发现,CIB2基因保守突变(p.E64D)导致CIB2切换到Mg2+结合形式的能力受损,从而导致USH1J综合征,而Booth等[25]认为CIB2中的双等位基因LOF突变导致常染色体隐性非综合征型聋(autosomal recessive non-syndromic hearing loss,ARNSHL)而不是USH综合征,提示CIB2基因突变与USH综合征的相关性有待进一步验证。
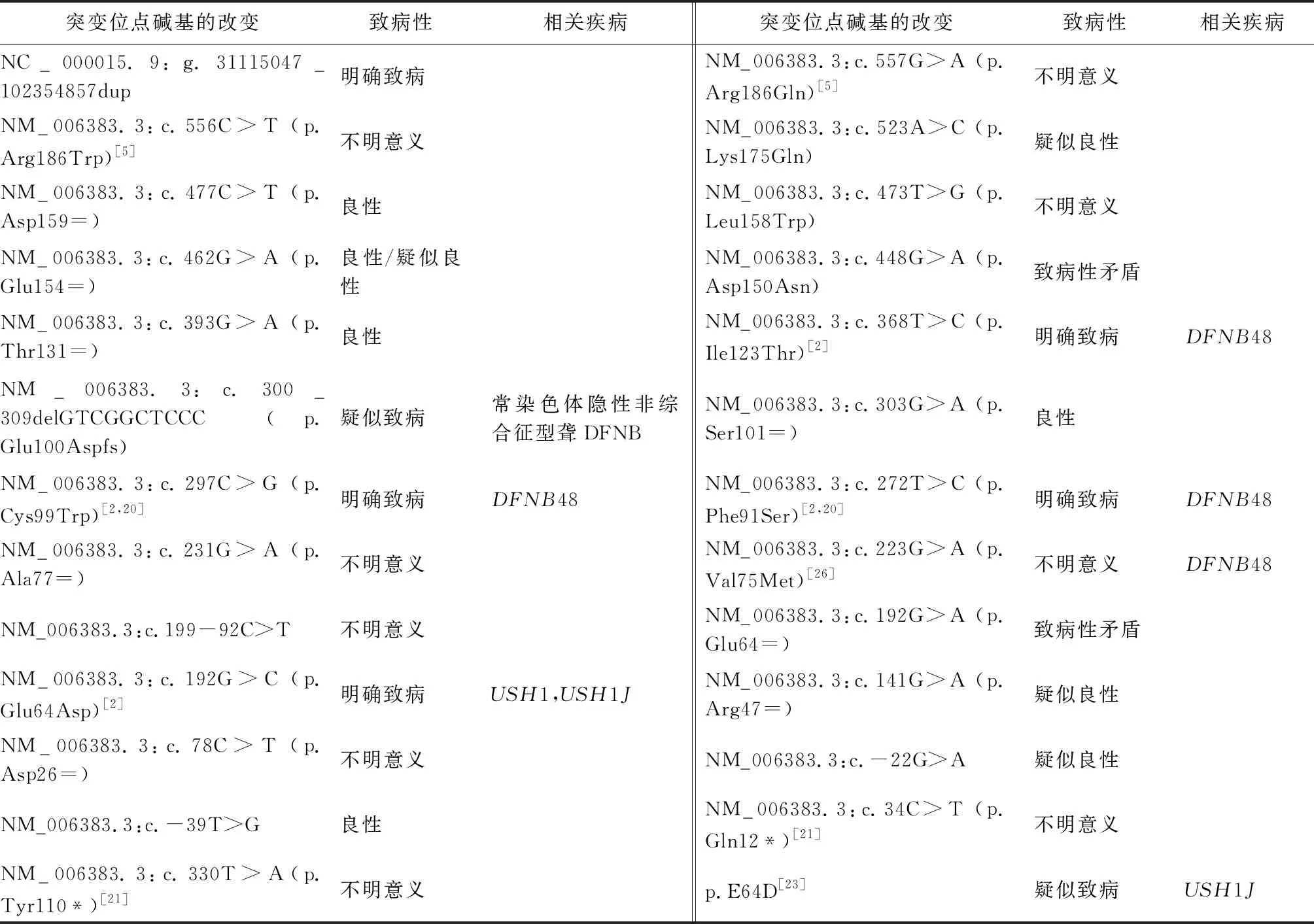
表1 CIB2基因耳聋相关突变位点碱基的改变及致病预测
注:表中记录均为CIB2基因与耳聋相关突变,转录本为NC_000015.9、NM_006383.3,其中除特别标注记录来源,数据参考均来自Clinvar数据库,致病性划分依据ACMG指南标准。

2.2CIB2基因突变致病机制预测
2.2.1CIB2基因突变破坏对Ca2+反应的抑制作用(图4)CIB2基因c.556C>T(p.Arg186Trp)突变破坏了异源表达系统中CIB2对ATP诱导的Ca2+反应的抑制,导致听力损失[5]。c.297C>G可破坏Ca2+效应结合位点,与之相反的是,c.368T>C可能放大了Ca2+的亲和力。细胞内Ca2+浓度对MET通道适应、频率调谐、发束抽搐、外毛细胞(OHC)、电活动信号和传入突触传递至关重要[2,27]。
2.2.2CIB2基因突变影响整合素亲和力及活性(图1,4)CIB2基因c.297C>G、c.192G>C突变改变了蛋白质构象,从而影响整合素的亲和力和活性,272T>C错义突变,使CIB2基因与整合素ɑIIβ的结合位点发生改变。整合素α7β1是主要的层粘连蛋白(laminin)α2链结合受体之一,负责适当的肌肉功能,Laminin2是肌肉发育和稳定性所必需的蛋白质[28],CIB2基因突变导致α7B亚基的表达减少、整合素活性降低[19]。整合素除了在邻近细胞间与ECM配体或受体的反作用之外,还用作从细胞膜外突触到膜内骨架的跨膜机械连接,该连接基于肌动蛋白的微丝系统[17]。
2.2.3CIB2基因突变影响与旋转蛋白和肌球蛋白VIIa相互作用(图4)CIB2基因c.192G>C突变导致USH1J综合征,USH的许多成员与肌球蛋白VIIa和旋转蛋白结合[2]。CIB2基因突变可能通过影响静纤毛相关蛋白(旋转蛋白Whirlin)从而调控静纤毛的发育。

3 CIB2动物模型研究
理解并阐明耳聋等疾病的细胞和生理机制对于临床治疗工作非常重要,而动物模型研究对于了解每一种疾病及研发更好的诊疗手段至关重要。小鼠、斑马鱼、果蝇等是研究视觉和听力损伤的较好的动物模型,也是分析哺乳动物发育、生理和疾病过程的常用模型动物。CIB2基因结构功能及致病性等研究的动物模型主要是小鼠、斑马鱼和果蝇等[12],其他,如:大鼠、绵羊,主要是CIB2蛋白定位研究的动物模型,如:CIB2蛋白定位研究发现其存在于大鼠的感觉、内嗅、前额皮质以及神经纤维中[15]和绵羊的胃、心脏和卵巢中[29]。
斑马鱼视觉和前庭声学系统组成与人类相似,且视觉和听觉系统发展迅速,受精后5天就能正常工作。斑马鱼视网膜含有典型的脊椎动物的光感受器、神经元、胶质细胞以及视网膜色素上皮(RPE)等结构。尽管其没有耳蜗,斑马鱼内耳支持细胞和毛细胞的组织和形态也可以与其他脊椎动物相媲美。斑马鱼的整个发育过程中均检测到CIB2,斑马鱼CIB2敲低模型研究发现,抑制斑马鱼中CIB2的表达会破坏斑马鱼对听觉刺激的反应且导致运动平衡失常及发育缺陷[2]。
CIB2小鼠模型的研究证实,CIB2基因的转录本存在于小鼠胚胎和出生后的整个发育过程中[2],如:内耳支持细胞、内毛细胞、OHC、角质板(肌动蛋白细胞形成的毛细胞顶端细胞质)和静纤毛细胞质中均检测到CIB2转录本,且出生后首先在Corti发育器官和前庭器官观察到CIB2转录本[2,22,30];在视网膜感光器、色素上皮细胞、神经节细胞等中也检测到CIB2转录本[2]。Riazuddin等[2]在2012年进行的小鼠体内CIB2基因敲除实验中发现,小鼠CIB2基因敲除影响了毛细胞尖端机械敏感传导(MET)和内耳立体纤毛的发育,并导致严重的听力损失;与CIB2基因敲除小鼠一样,所有发生CIB2突变小鼠都表现出严重的听力损失。两个独立科研小组在2017年相继报道了CIB2敲除小鼠出现重度聋,该研究还显示CIB2可结合TMC1/2,在CIB2敲除小鼠毛细胞中完全不存在MET电流[31,32],TMC蛋白是一种典型的MET通道蛋白基因[22]。
果蝇基因CG9236,其编码的蛋白与人CIB2基因具有59%的同一性,果蝇模型研究可以进一步评估CIB2功能。CG9236(dCib2)在幼虫和成虫多种组织中表达,包括成虫眼中。在果蝇dCib2 RNAi敲低实验中发现,敲低dCib2 RNAi果蝇出现显著降低的光响应幅度以及对闪烁刺激的响应受损,该结果还表明dCib2是实现强烈、持续的光响应和跟踪快速光刺激所必需的[2]。
4 小结与展望
目前已报道CIB2基因突变有四十多种,其中与耳聋相关的突变达二十多种,越来越多研究表明CIB2基因已成为遗传性聋的主要致病基因之一。近期多篇报道质疑CIB2突变导致USH1J综合征的准确性,其具体的致病机制有待进一步研究阐明。CIB2与MET通道的关系及其在哺乳动物听觉系统中的功能、CIB2与旋转蛋白和肌球蛋白Vlla相互作用的具体机制、CIB2突变致聋的生理机制等仍不清楚,需要加强对CIB2基因结构及功能等的基础研究,以便更清楚的了解和预测其突变的致病机制和致病性。高通量测序、基因编辑等实验技术的高速发展将加快CIB2基因研究的速度,而我国人群中CIB2基因突变的突变型及其致病性和分布特点等尚无系统的数据报道,未来可充分利用高通量测序技术等对我国人群中耳聋患者和正常人群进行CIB2基因捕获测序并进行相关统计学分析及数据库建立,以便更精准有效地预防和治疗耳聋、心血管和乳腺癌等疾病,为USH综合征等聋病的产前诊断、遗传咨询和基因治疗提供可靠的数据支持。
猜你喜欢
医学综述(2022年10期)2022-11-28
安徽医科大学学报(2022年6期)2022-07-13
小天使·二年级语数英综合(2021年8期)2021-08-16
听力学及言语疾病杂志(2020年4期)2020-12-20
口腔医学(2020年6期)2020-12-19
复旦学报(医学版)(2020年2期)2020-04-17
电脑报(2020年4期)2020-03-25
作文评点报·作文素材小学版(2016年8期)2016-03-16
第二课堂(小学版)(2015年2期)2015-05-22
癌变·畸变·突变(2015年3期)2015-02-27
