
SO42-/ZrO2固体超强酸催化剂的制备及其对正己烷异构化反应的影响研究*
2020-05-19 03:19艾罡
陶瓷 2020年2期
艾 罡
(榆林康耐雅新材料科技有限公司 陕西 榆林 718100)
前言
烷烃异构化反应为微放热反应(2~20 kJ/mol),故低温有利于反应的进行。但从动力学角度看,提高温度有利于反应进行。C5/C6异构化按操作温度可分为高温异构化(高于320 ℃)、中温异构化(200~320 ℃)和低温异构化过程(低于200 ℃)3种,其中高温异构化现已基本淘汰。国外C5/C6异构化工艺开展很早,已有近100套装置运行或在建。低温型异构化催化剂以卤化铂/氧化铝为代表,低温活性高,热力学上允许较多的支链产物生成,但催化剂易被微量水和硫中毒;中温型异构化催化剂以铂/改性丝光沸石为代表,需在较高温度下(250~280 ℃)操作,热力学平衡转化率低,但催化剂抗中毒能力强,具有很强的竞争力。
SO42-/ZrO2型固体超强酸具有催化活性高、选择性强、制法简单、稳定性好、不腐蚀设备、不污染环境等特点,是对环境友好、很有应用前景的绿色工业催化剂,被广泛应用于裂解、异构化、烷基化、酯化、酸基化、聚合等反应[1~2]。关于SO42-/ZrO2型固体超强酸催化活性中心,考虑到Bronsted酸位的形成,这种模型基于这样的假设——认为表面的硫化物是以硫酸氢根离子的形式存在,它在化学吸附的时候能和Zr-OH-Zr产生键桥作用;当加热时,硫酸氢根离子能和相邻的OH作用,或是两个相邻的OH能相互反应,前者导致产生Lewis酸位,后者导致产生Bronsted酸位[3]。
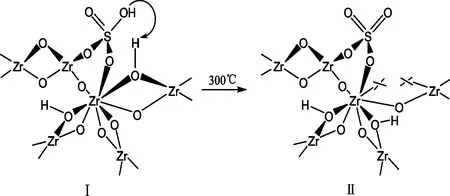
图型酸位形成的假设模型
由于超细Zr(OH)4具有比表面积大、粒径分布范围比较小的特点,所以在催化剂的负载过程中容易得到大比表面积的、活性位比较丰富的催化剂[4]。笔者的研究内容主要是考察金属氧化物改性SZ固体超强酸催化剂的正己烷异构化的研究。
1)通过改变硫和铂的浸渍比例、焙烧温度等制备条件,制备各类PSZ、PSZA催化剂。
2)摸索此催化剂的反应条件,如焙烧温度、活化温度、还原温度等,对正己烷异构化反应活性的影响,以便充分发挥催化剂反应性能。
3)为了克服SZ催化剂容易失活和酸性强度不够的缺点,向催化剂体系引入贵金属、Al2O3、稀土La2O3、Ce2O3、YeO的改性。摸索各种改性合适的引入方式,包括成形法、共沉淀法、浸渍法等,以达到促进SO42-/ZrO2催化剂的性能。
4)比较Al2O3、La2O3、 Ce2O3和YeO3分别改性SO42-/ZrO2催化剂在晶型、焙烧过程、比表面积、酸性、正己烷反应性能等方面的差别,得出相应规律,以便指导催化剂的制备工艺。
1 实验部分
1.1 实验仪器与试剂
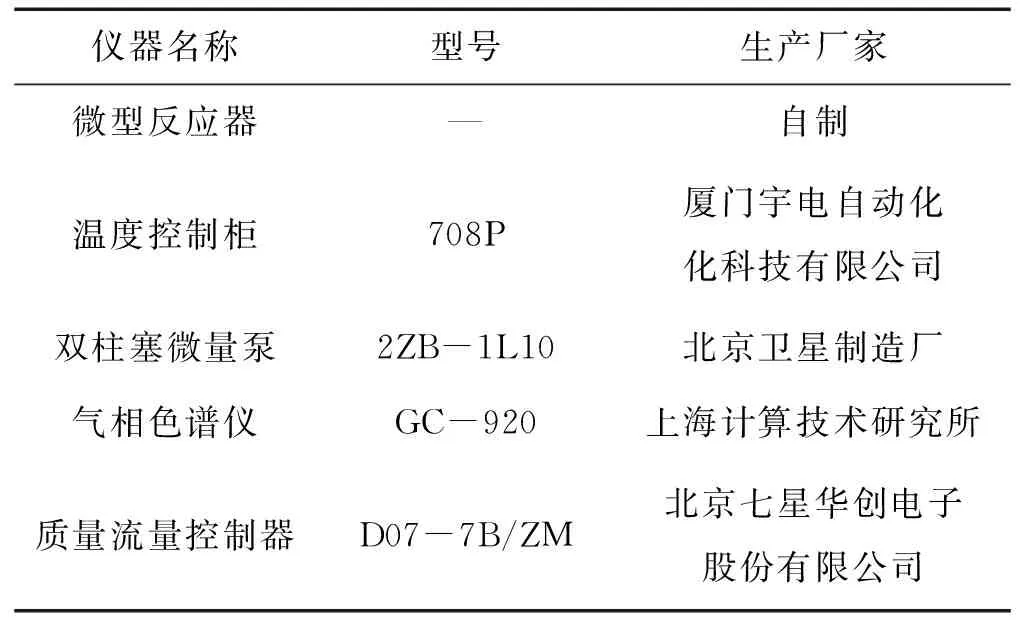
表1 实验主要仪器
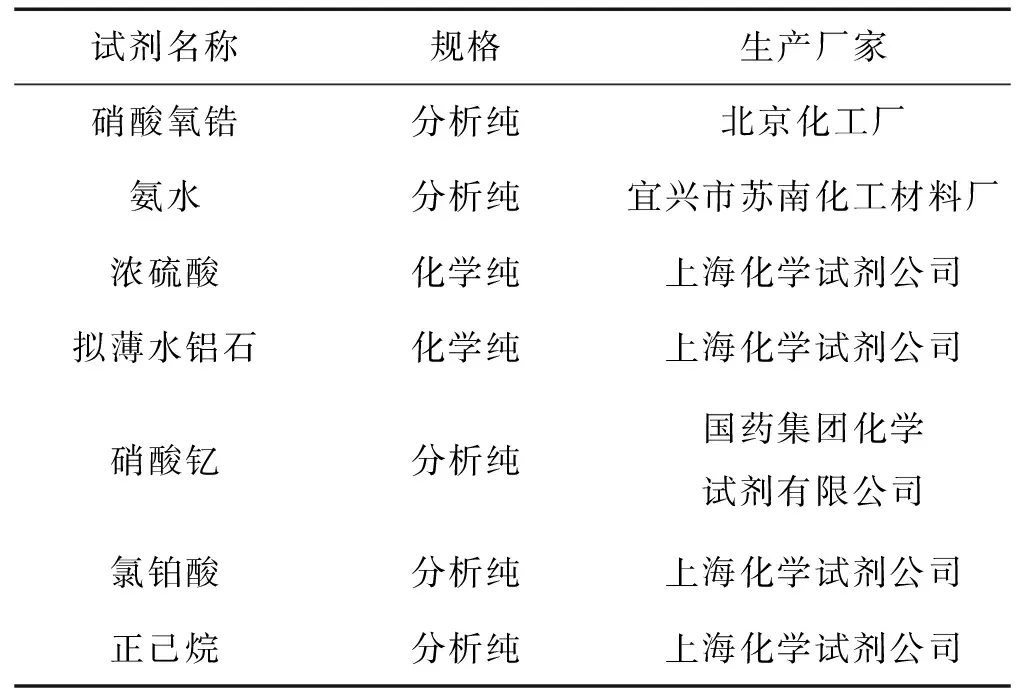
表2 实验主要试剂
1.2 催化剂的制备及表示方式
1.2.1 水合氧化锆的制备
水合氧化锆载体的制备采用直接沉淀法:采用了酸碱同时滴加的方法,在把含[Zr4+]为0.4 M的硝酸氧锆滴入已配好的氨水溶液中,同时在强烈搅拌的情况下用分液漏斗滴加一定量的浓氨水,保持pH值>10。具体做法为:
1)用500 mL去离子水溶解53.454 g硝酸氧锆固体。
2)量取100 mL氨水,并用去离子水稀释到1 000 mL。
3)在大烧杯中留500 mL稀释后的氨水,其余的氨水与硝酸氧锆溶液以1∶1的速度滴加到装有500 mL稀释后氨水的大烧杯中,边滴加边搅拌。滴加完毕后持续搅拌1 h,将其静止老化过夜后,用去离子水将所产生的白色沉淀洗涤至中性,抽滤,110 ℃干燥12 h,研磨备用。
1.2.2 SO42-/ZrO2(SZ)的制备
上述方法制备的水合氧化锆为载体,用0.5 M的硫酸溶液浸渍5 h,液固比为15 mL/g。抽滤后110 ℃干燥12 h,再在马弗炉中650 ℃焙烧3 h后得到SO42-/ZrO2,样品分别记作SZ和SZt。
1.2.3 助剂的引入
1)助剂Al的引入:称取一定量直接沉淀法制备得到的Zr(OH)4粉末和拟薄水铝石(最终Al2O3/ZrO2=5/100),滴加浓度为15%的稀硝酸溶液混合均匀后,挤条成形,110 ℃干燥12 h后,载硫,再同2.2.3b中方法载Pt,得到样品记为PSZA。
2)助剂Pt的引入:以一定浓度的H2PtCl6溶液浸渍上述制备的SO42-/ZrO2(铂含量为0.5wt%),室温下静置24 h后,于110 ℃干燥12 h,最后在马弗炉中500 ℃焙烧3 h,最终得到样品分别记为PSZ和PSZt。
3)助剂Y、C、L的引入:以直接沉淀法制备得到的Zr(OH)4粉末,载硫并干燥后,分别用硝酸钇、硝酸铈、硝酸镧溶液于室温下浸渍上述载硫的母体以引入稀土元素Y、C、L,其摩尔浓度为3%mol,10 h后在110 ℃下干燥12 h,再在马弗炉中650 ℃焙烧3 h后,载Pt,得到样品记为PSZY、PSZC、PSZL。同法在引入助剂Al的基础上得到的样品记为PSZAY、PSZAC、PSZAL。
1.3 催化剂的活性评价
正己烷临氢异构化反应是在不锈钢微型反应器(内径为5 mm)中进行的,催化剂装填量为2 g,装填方式为上下端以石英砂,并用石英棉隔开分层填充,反应管中间为催化剂床层。具体反映操作步骤为:
1.3.1 活化
反应前,催化剂先在350 ℃下通空气活化3 h。
1.3.2 还原
活化完毕后,降温至250 ℃在H2气氛(氢气压力为0.5 MP)下还原1 h。
1.3.3 反应
还原完毕后,降至所需反应温度。反应在临氢条件下进行,WHSV为1h-1,氢油比为10,反应压力为2.0 MPa。反应器上端进料,下端出料。氢气从高压钢瓶经减压净化干燥后进入反应器,通过氢气流量计控制氢气流速,反应物料由2ZB-1L10型双柱塞微量泵输送,流量稳定。反应尾气由微调阀调节流量后放空,反应产物由六通阀取样后进入色谱系统进行在线分析。整个反应工艺流程的管路用3 mm×0.5 mm的不锈钢管连接,实验中所用色谱为上海计算技术研究所生产的GC-920型气相色谱仪,其毛细管色谱柱柱长为30 m,以OV-101为固定相,氮气作为载气,产物由氢焰离子检测器(FID)检测,产物分布由S100色谱工作站处理按面积归一法定量。
1.4 催化剂物化性能表征
1.4.1 程序升温还原(TPR)
程序升温还原(TPR)在Auto Chem II(Micromeritics,USA)上进行,催化剂样品在含9.16%氢气的氩气气氛中程序升温还原,气体流量分别为30 mL/min,升温速率10 ℃/min,温度范围为40~900 ℃。氢气消耗量使用仪器自带的热导检测器(TCD)测量。
1.4.2 NH3-程序升温脱附(NH3-TPD)
用NH3-TPD法测定催化剂的表面酸性。以He气为载气,流量18 mL/min,催化剂装量0.14 g,颗粒大小为20~40目。样品在600 ℃下活化30 min,然后冷却至150 ℃,吸NH3至饱和,经He吹扫除去物理吸附的氨后,以18 ℃/min升温速率进行脱附至700 ℃,脱附的NH3用热导检测。
2 实验结果与讨论
2.1 硫酸锆的焙烧温度对催化剂异构化活性的影响
对于硫酸化后的氢氧化锆催化剂,焙烧是制造有活性催化剂的必要步骤。图2为焙烧温度对PSZA催化剂异构化活性的影响。从图2可以看出,随着硫酸锆的焙烧温度由575 ℃升高到625 ℃,PSZ的异构化活性明显增加,二甲基的收率也有所增加;继续升高焙烧温度,异构化活性明显下降,二甲基的收率也随之下降。对于上述在不同温度焙烧硫的催化剂,正已烷在其上200 ℃反应时,仅有轻微裂解发生,其异己烷的选择性均在90%以上。这与张黎等[5]的报道相一致,为了产生强酸中心硫酸锆的焙烧是必要的,但焙烧温度过高,催化剂上的硫会因分解而流失,是导致催化剂活性下降的可能原因。由此可见,硫酸锆较为适宜的焙烧温度为625 ℃。
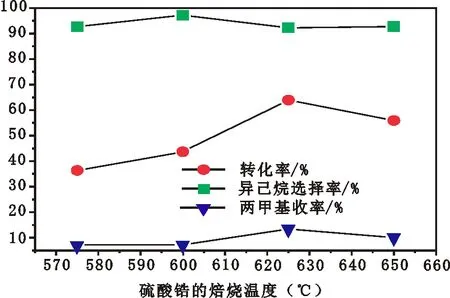
P=2.0 MPa;H2/n-C6=10∶1;Reaction Temperature=200 ℃,WHSV=1图2 硫酸锆的焙烧温度对PSZ催化剂异构化活性的影响
2.2 铝含量对PSZA催化剂活性的影响
最初在SO42-/ZrO2固体超强酸中加入氧化铝,只是为了解决催化剂的成形问题,但却发现氧化铝的引入使催化剂的活性也有了显著提高[6]。笔者考察了铝的加入对PSZ催化剂的异构化活性影响,结果如图
3所示。从图3可知,铝的加入明显影响了催化剂的异构化活性。PSZ上的异构烷烃收率约为60%,3%铝的添加使得异已烷的收率提高到65%左右,当铝含量增加到5%时,PSZA5上的异己烷收率与未添加铝的PSZ上的异己烷收率相当,继续提高催化剂中的铝含量,异己烷收率进一步下降,继续增加铝含量到20%时,正己烷的转化率非常低,仅为15%左右。可见,加入铝显著影响了催化剂的异构化活性,且适宜的铝含量为3%。而夏勇德等[7]的研究结果表明,铝的添加大大提高了SO42-/ZrO2催化剂的异构化活性,而对铂改性的SO42-/ZrO2的催化剂体系而言,铝的加入没有明显提高PSZ的异构化活性。这与本次研究的结果有所不符。已有大量的实验结果显示,对于SO42-/ZrO2固体超强酸烷烃异构化体系,催化剂吸附的水对其异构化活性有一定的影响,而铝的加入又会影响催化剂对水的吸附。由此推断,两者结果的差异可能与催化剂上吸附水的量不同有关,进一步考察其的研究正在进行中。
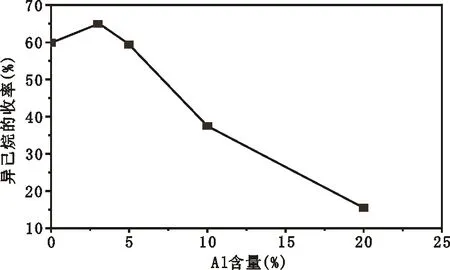
P=2.0 MPa;H2/n-C6=10∶1;Reaction Temperature=220 ℃,WHSV=1图3 不同铝含量的PSZA催化剂的异构化性能
2.3 Al添加方式对催化剂异构化活性的影响
SZ催化剂的活性很大程度上取决于其制备过程,上节讨论了铝含量对PSZ催化剂异构化活性的影响,在铝含量一定的条件下,铝的添加方式也可能为影响催化剂活性的一个重要因素,本节考察铝的添加方式对催化剂异构化活性的影响(铝的质量百分数为3%),具体如表3所示。
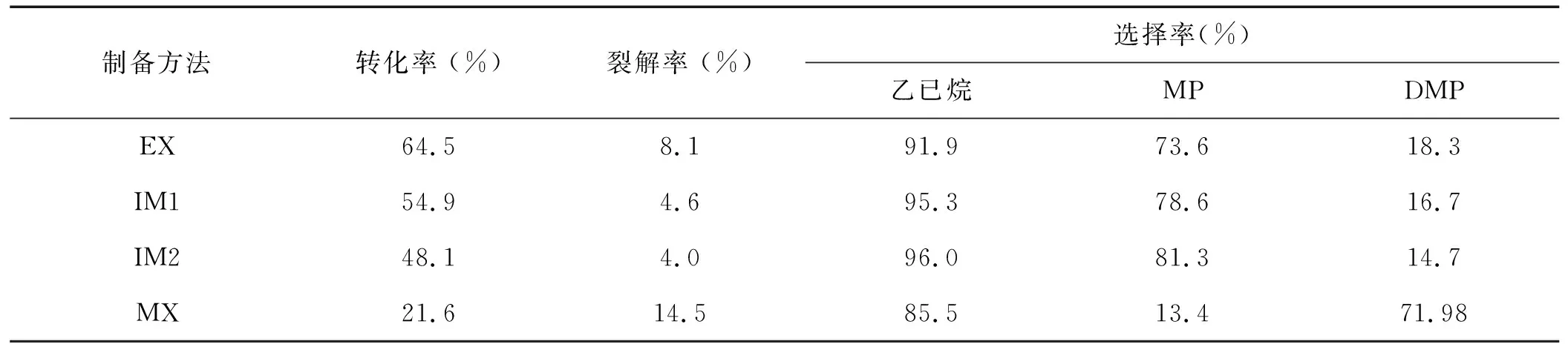
表3 铝添加方式不同的催化剂样品的异构化活性
由表3可见,不同方法引入铝的催化剂异构化活性差异较大。通过浸渍载铝后挤条成形法(EX)制备的PSZA催化剂上的正己烷转化率最高,为64.5%,异己烷的选择性也在90%以上。而通过机械混合法(MX)制备的PSZA催化剂的正已烷转化率和选择率都是最低的,分别为21.6%和85.5%,但这种催化剂DMP选择率却高达71.98%,远远高于其它方法制备的催化剂的DMP选择率。对于用浸渍法载入铝所制得的PSZA(IM)催化剂,载硫后用浸渍载铝标记为PSZA(IM1),载硫前浸渍的标记为PSZA(IM2),这两种催化剂的异已烷转化率和DMP选择率都居于前两种催化剂中间,且催化性能相差不大,说明载硫和载铝的前后顺序对催化剂的催化性能影响不大。由表还可以看出,两种用浸渍法载铝制得的PSZA催化剂的裂解产物很少,均不高于5%。
反应条件:220 ℃,1 h-1,2.0 MPa,n(H2)/n(hexane)=10 (MP代表2-methyl pentane and 3-methyl pentane;DMP代表2,2-dimethyl butane and 2,3-dimethyl butane) EX:挤条;IM1:硫酸化后的Zr(OH)4再浸渍Al2SO4IM2:含Al2SO4的硫酸溶液浸渍Zr(OH)4,MX:机械混合。
由于机械混合法(MX)制得的PSZA催化剂活性很差,所以这里重点对其它3种载铝的方法对催化剂晶相结构和热分析进行讨论。晶相结构由XRD测定如图4所示。
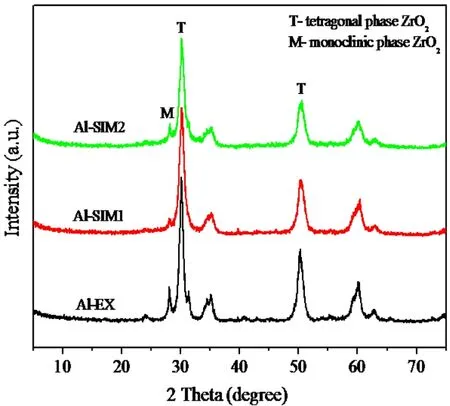
图4 不同铝添加方式制备的PSZA的XRD图
由图4可以看出,3种催化剂均是以四方相为主,IM1与IM2的晶相结构几乎没有区别,即铝的添加顺序对其晶相结构无影响。而用挤条成形法(EX)载入铝的PSZA催化剂的单斜相稍比浸渍法载铝的多,四方相几乎没有区别。通常认为四方相氧化锆是硫酸锆基催化剂的活性相,SO42-必须以一定形式与四方相ZrO2结合才能形成超强酸[8]。因此,3种催化剂均具有较好的活性,但是,EX法载铝的PSZA催化剂比IM法催化剂的单斜相多,活性却比它的活性高,这可能因为载铝的不同方式对催化剂结构的微小影响,对其异构化活性关联不大,载铝方式对催化剂本质的影响来自于其它方面[9]。
再对催化剂进行TG表征进行热分析,即图5。由图5可见,催化剂的失重主要分为2个阶段。低于500 ℃的失重主要是催化剂表面吸附水的脱除,500 ℃以上的失重与催化剂上的硫酸根分解有关。图中的数字为各样品的硫含量,其计算公式为:
SO42-amount (g/g cat) = (M1-M2)/ M1
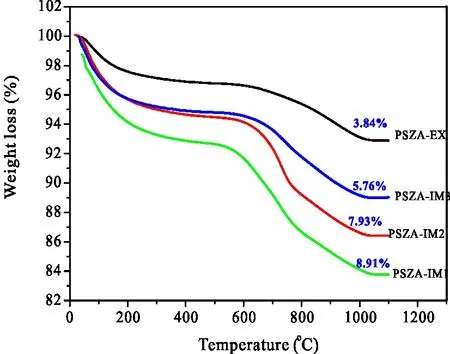
图5 不同方法加入铝的PSZA的TG曲线
挤条成形法(EX)制得的PSZA催化剂的硫含量最少,仅为3.84%;IM1的最多,为8.91%;IM2的居中,且与IM1的相差不大,仅有1%左右。可以看出,催化剂载铝方式不同,各催化剂的硫含量有较大区别,Al的添加顺序对硫含量的影响也不是很大。将催化剂的硫含量与其异构化活性相联系可得,催化剂上的硫含量与其异构化反应活性也不能很好的关联,较高的硫含量并非获得高异构化活性的决定因素,可能还与硫在催化剂上的分布有关。综上所述,用浸渍载铝后挤条成形(EX)法制备的PSZA正已烷异构化催化剂的异构化活性最佳。载铝方式的不同导致催化剂晶相结构的变化,并不是影响其活性的主要因素;催化剂上硫含量并不与其异构化活性成正比,即较高的硫含量并非意味着高活性,可能与其在催化剂上的分布有关。
2.4 助剂C、Y和L的添加PSZ催化剂活性的影响
不同稀土元素改性的PSZ催化剂正己烷转化率如图6所示。从图6可见,PSZ催化剂催上的正己烷异构化活性最高,PSZC和PSZY次之,PSZL最低。由此可见,引入稀土元素并没有提高PSZ催化剂的正已烷异构化活性。
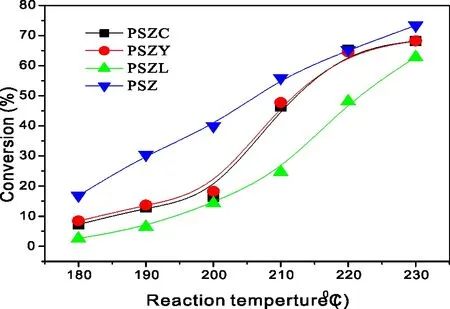
P=2.0 MPa;H2/n-C6=10∶1;WHSV=1
图7为对不同催化剂进行晶相结构表征的XRD谱图。由图7可以看出,在PSZ催化剂以四方相为主,存在有少量的单斜相,而稀土改性后的催化剂,单斜相均消失,均呈现出四方相。说明稀土的引入,有利于催化剂由单斜相向四方相转变。将催化剂的晶相结构与其异构化活性相关联,与2.3节的结果相类似,助剂的加入导致晶相结构的变化对其异构化活性影响不大。
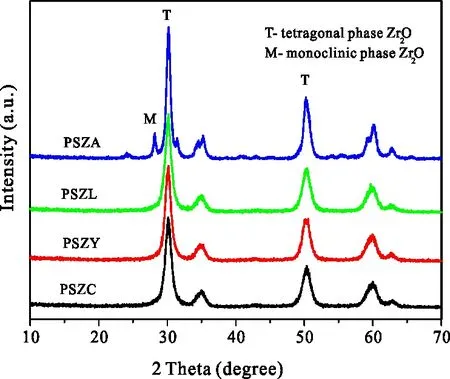
图7 不同稀土改性的PSZR催化剂的XRD图
图9为对不同催化剂进行H2-TPR表征测其还原性,结果显示,PSZ及稀土改性后的PSZ样品TPR曲线均出现一个明显的还原峰,没有添加助剂的PSZ催化剂还原峰位于550 ℃,助剂改性的PSZ催化剂的还原峰温度大致相同,向低温方向移动约50 ℃,即出现在500 ℃左右。由此可见,助剂的加入降低了催化剂上的硫在还原气氛中的稳定性,这与Davis L等[10]的结果相一致,即助剂的加入使得硫酸根更易被还原。
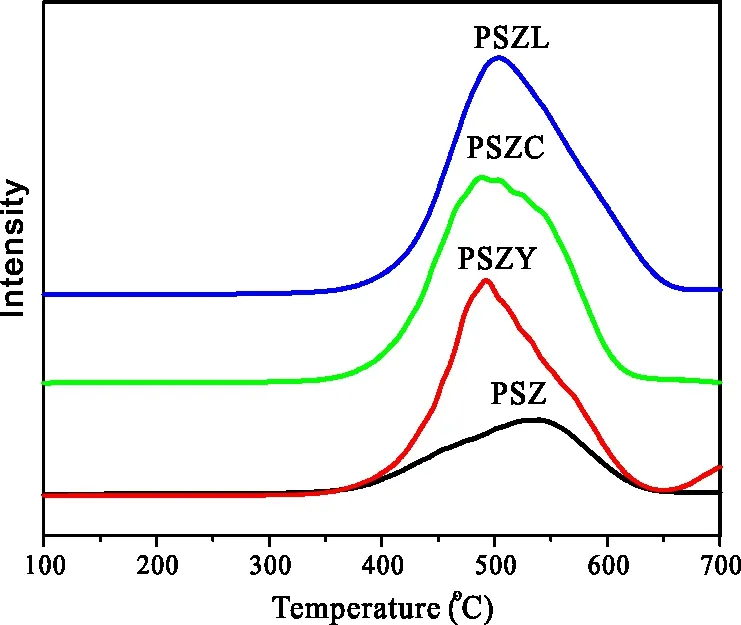
图8 不同催化剂的H2-TPR图
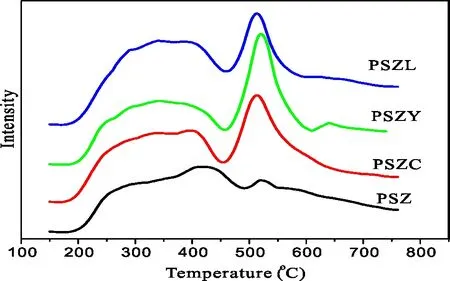
图9 不同催化剂的NH3-TPD图
不同催化剂的酸性通过NH3-TPD表征,结果如图5、图6所示。脱附温度代表酸的强度,脱附峰的面积代表酸中心的数量。低于450 ℃的脱附峰代表弱酸中心,高于450 ℃的脱附峰则与强酸中心有关。从图5、图6可以看出,样品PSZ、PSZC、PSZY和PSZL均在350 ℃和510 ℃出现两个脱附峰。这表明4种样品分别具有一定数量的弱酸和强酸中心。观察脱附峰出现的温度和脱附峰的面积,4种催化剂样品酸强度大致相同,而酸量由高到低顺序为PSZY>PSZC>PSZL>PSZ。由此可见,助剂C、Y和L改性过的PSZ催化剂酸强度的变化并不明显,但酸中心的数量随着助剂加入有明显增多。
将正已烷转化率与其H2-TPR、NH3-TPD表征结果相关联,因其反应温度(小于230 ℃)并没有达到H2-TPR表征得到的还原峰出现的温度,因此认为催化剂样品上的硫,在反应过程中没有被还原,对反应的影响可以忽略。但与NH3-TPD表征结果不相符这一点,目前还不能很好的理解,初步认为可能是NH3-TPD表征环境和反应环境不相同,在不同的环境下,催化剂样品的酸中心数量和强度可能会发生相应改变,从而导致表征结果与实验数据出现了一定偏差。
2.5 还原温度的的影响
还原温度对PSZA催化剂异构化活性的影响如图10所示。
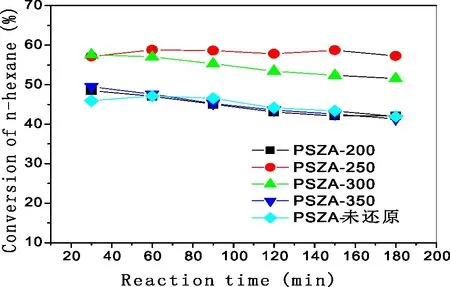
P=2.0 MPa;H2/n-C6=10∶1;Reaction Temperature=200 ℃;WHSV=1图10 还原温度对PSZA催化剂正己烷异构化活性的影响
在不同还原温度下,PSZA催化剂的正己烷转化率表现出明显差异,正己烷转化率由大到小顺序为PSZA-R250>PSZA-R300>PSZA-R200(PSZA -R350、PSZA-未还原),即250 ℃下还原有最高的异己烷收率。还原温度对异己烷的选择性影响甚微。若催化剂未还原,其活性与200 ℃和350 ℃还原的催化剂异构化活性相当,这可能是催化剂在进行焙烧后,已经有一部分铂以金属态附着在催化剂上,这部分铂不需要经过还原,便能对反应起到催化的作用,从而提高催化剂的异构化活性。但催化剂若未经焙烧就进行反应会有一定的诱导期。随着还原温度的升高,金属态的铂随之增加。由图9H2-TPR表征结果可知,脱附峰始于300 ℃以上,因此温度低于300 ℃下的还原,不会导致催化剂上硫物种的损失,其影响可不考虑。但当还原温度高于300 ℃时,催化剂上的硫物种会被还原为H2S而损失,催化剂活性下降,即300 ℃以上的还原温度对催化剂活性的影响不可忽略。结合反应结果,与250 ℃还原相比,300 ℃下还原的催化剂具有较低的活性。虽然300 ℃下还原并不能导致催化剂上硫的损失,但硫的化学状态可能发生变化。通常认为硫酸锆基催化剂中起活性作用的硫物种为高价态的硫,而300 ℃下还原可能导致部分高价态的硫转变为低价态,从而导致活性的下降。进一步提高还原温度到350 ℃,部分硫被还原为H2S而损失,催化剂活性进一步下降[12]。
2.6 PSZ和PSZA催化剂放置时间及吸水对异构化活性的影响
PSZ、PSZA催化剂放置时间及吸水对异构化活性的影响如图11和图12所示。从图11和图12可见,放置时间不同,PSZ(PSZA)催化剂的异己烷的收率有差异,2种催化剂均是未放置即原位焙烧的异构化活性最低,放置5 d时正已烷转化率最高。但是若催化剂放在密封装有水的烧杯中使其吸水,其活性比未放置的催化剂还要低。对PSZ催化剂,吸水3 d后的活性为22%,原位焙烧的活性约为40%,放置5 d时活性为62%,正已烷转化率提高了20%,而进一步增加放置时间至20 d,正已烷转化率又略有下降,低于60%。对PSZA催化剂,催化剂活性随放置时间变化的趋势和PSZ催化剂的相一致,即催化剂放置3 d吸水的活性最低,然后是原位焙烧的催化剂,再就是放置2 0d,放置5 d的催化剂活性最高。同样的催化剂,活性随放置时间不同的差异,与将其放在饱和的水蒸气环境中使其吸水均与催化剂中的含水量有关。催化剂在放置过程中不可避免地会吸附空气中的水,将其放在装有水的烧杯中吸水3 d使其吸水饱和。这均是考察水对催化剂活性的影响。前面已有许多学者发现了水对催化剂活性有一定的影响。水对催化剂活性起抑制作用,但也有学者的实验结果却显示水对它起促进作用[13~14]。这两者的矛盾可能为水对催化剂活性的影响与其吸水量有关。适当的吸水量对催化剂活性起促进作用,过多或过少均起抑制作用,这与实验结果也几乎一致,催化剂活性随放置时间变化有先增大再减小的趋势,将其放在密封的环境中吸水后,水对催化剂活性的影响更大。比较图11和图12可以看出,PSZA催化剂吸水3 d后活性为34%(放5d时活性为67%),PSZ催化剂吸水3 d后活性为22%(放5 d时活性为62),说明铝的加入,提高了PSZ系列催化剂耐水性。综上所述,催化剂上的吸附水对其异构化活性影响很大,且对不同的催化剂,水对它的影响也有一定的差别。由于实验条件的限制,不能将催化剂的最佳吸水量进行定量表示,初步将其定为焙烧后再放置5d使其吸附有较适量的水,从而具有较好的催化活性。
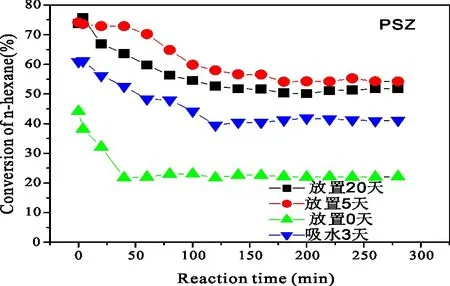
P=2.0 MPa;H2/n-C6=10∶1; Reaction Temperature=200 ℃;WHSV=1图11 PSZ催化剂放置时间及吸水对其异构化活性的影响
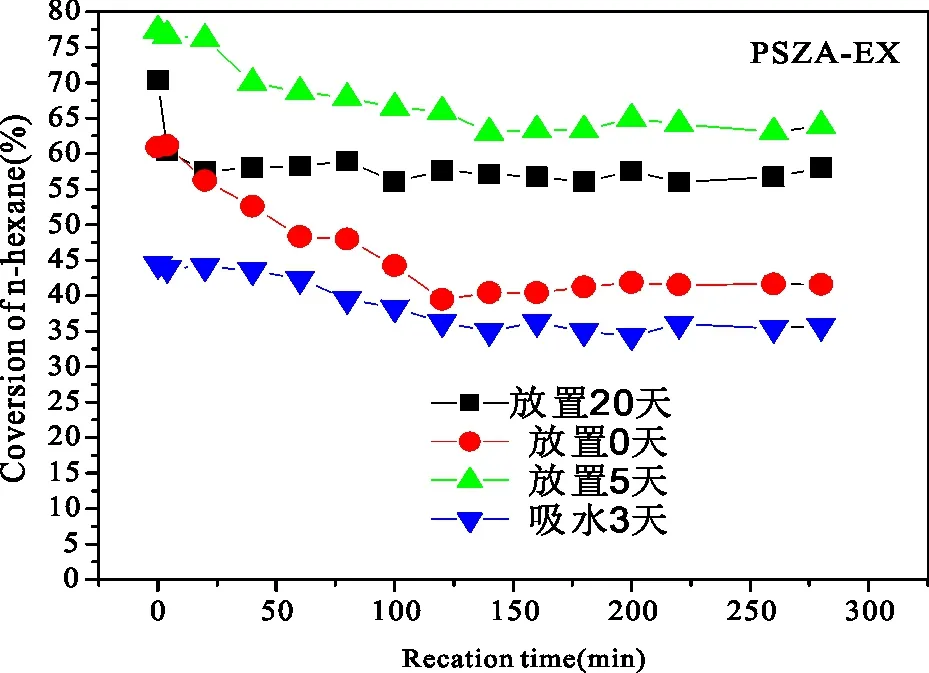
P=2.0VMPa;H2/n-C6=10∶1; Reaction Temperature=200 ℃;WHSV=1图12 PSZA催化剂放置时间及吸水对其异构化活性的影响
2.7 活化温度对催化剂异构化活性的影响
催化剂经过干空气活化,主要是消除催化剂上吸附的水与一些杂质。催化剂的活化温度对异构化活性的影响如图13所示。由图13可见,活化温度的变化导致了催化剂异构化活性的明显变化。不同活化温度下正己烷转化率由高到低顺序为PSZA-A350>PSZA-A250(PSZA-A430)>PSZA-A110。而不同活化温度下异己烷的选择性均在95%以上。上节已讨论了催化剂吸附的水对其异构化活性有影响,合适的吸水量有能促进异构化反应。本次研究所用的催化剂均为焙烧后放置在空气中,催化剂不可避免地会吸附空气中的水。活化温度的不同会导致催化剂上吸水量的不同。过低的活化温度(110 ℃)导致催化剂上仍吸附有大量水,从而抑制其活性,当活化温度过高,如450 ℃,又会导致催化剂上大量吸附水的脱除,此时活性不佳。M Signorettoyi[15]以及J M Kobe[16]两个课题组的研究表明催化剂上适量吸附水的存在大大促进了异构化反应的进行。在350 ℃下活化,催化剂含有较适量的吸附水,活性也较好。对PSZ系列催化剂,较适宜的活化温度为350 ℃。
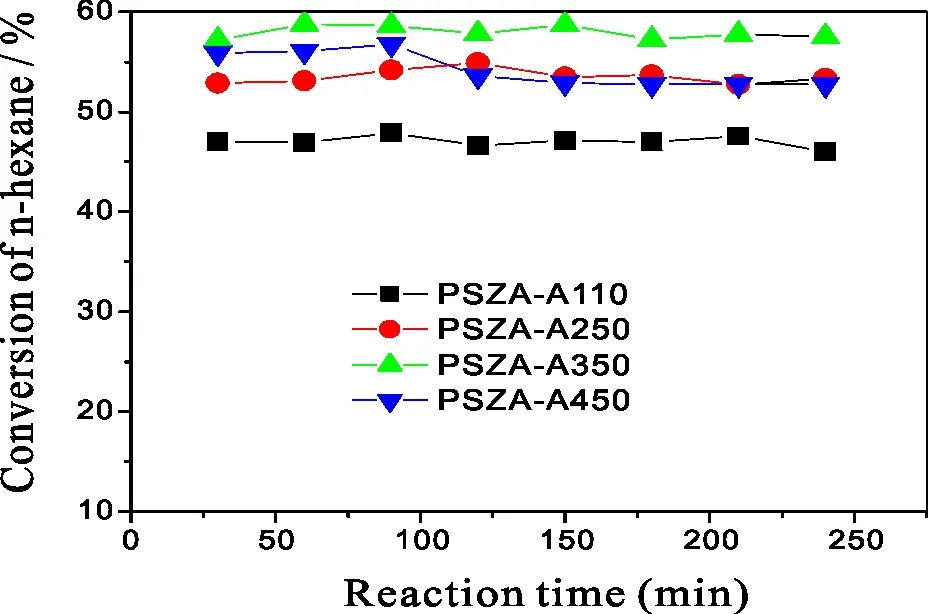
P=2.0 MPa;H2/n-C6=10∶1;Reaction Temperature=200 ℃;WHSV=1图13 不同活化温度催化剂的正己烷异构化活性
3 结论
1)高温焙烧为SO42-/ZrO2产生强酸中心的必要条件,但焙烧温度过高,催化剂上的S又会分解流失而导致活性下降。对PSZ正已烷异构化催化剂,SO42-/ZrO2的最佳焙烧温度为625 ℃。
2)氧化铝的加入除解决催化剂成形问题外,还显著提高了催化剂的异构化活性,较适宜的铝含量为3%。
3)SZ催化剂载铝有多种不同的方法,EX:挤条;IM1:硫酸化后的Zr(OH)4再浸渍Al2SO4。IM2:含Al2SO4的硫酸溶液浸渍Zr(OH)4。MX:机械混合等方法。经实验结果分析硫酸化后的Zr(OH)2再挤条添加Al元素的方法是提高PSZ催化剂异构化活性的一个很有效的方法。因此用挤条的方法添加铝是提高催化剂性能的一个很好的办法。载铝方式的不同导致催化剂晶相结构的变化,并不是影响其活性的主要因素;催化剂上硫含量并不与其异构化活性成正比。
4)由XRD表征,助剂的引入有利于单斜相向四方相的转变,但这所引起的异构化活性并没有提高催化剂的异构化活性,说明助剂的引入导致的晶相结构改变并不是影响其异构化活性的主要因素。通过NH3-TPD表征,助剂Y、C、L的引入增加了催化剂的酸中心数量,但由实验结果得出,添加助剂C、Y和L并不能改善PSZ催化剂的异构化活性。初步认为NH3-TPD表征环境和反应环境并不相同,很可能在反应过程中,催化剂样品的酸中心数量和强度与表征时得到的酸中心数量和强度发生了较大的变化。
5)还原步骤有利于铂以金属态附着在催化剂表面起加氢和脱氢的作用,从而增强催化剂的活性。但还原温度过高,又会导致催化剂上硫物种的损失而失活,实验结果表明,催化剂在250 ℃下还原有利于其反应。
6)催化剂的放置时间与活化温度,均通过影响催化剂的含水量来改变其活性。笔者的实验结果和一些文献报道的内容均显示,吸水量对催化剂的异构化活性有显著影响,合适的吸水量有利于促进异构化反应,过多或过少都会抑制催化剂的活性。经实验研究放置时间为5 d左右,活化温度为350 ℃可保持催化剂适量吸附水的存在,从而具有较高的异构化活性。
猜你喜欢
化工设计(2022年4期)2023-01-02
陶瓷学报(2021年1期)2021-04-13
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
今日农业(2020年20期)2020-11-26
石油石化绿色低碳(2020年1期)2020-04-08
电子技术与软件工程(2016年24期)2017-02-23
中学化学(2015年2期)2015-06-05
中国医药科学(2015年4期)2015-05-20
新课程·中学(2014年7期)2014-10-24
理科考试研究·高中(2014年8期)2014-10-17
