
Lyocell纤维纺丝原液稳定剂PBD的合成及应用研究
2020-05-15 01:10杨彦菊张慧慧杨革生邵惠丽
合成技术及应用 2020年1期
杨彦菊,彭 康,张慧慧,杨革生,邵惠丽
(纤维材料改性国家重点实验室,东华大学材料科学与工程学院,上海 201600)
Lyocell纤维作为生物基纤维新材料的代表,其在服装、家用、防火阻燃、抗菌医疗等领域使用比例逐年增高,并且,Lyocell纤维的生产过程无任何化学反应,对环境无污染,满足人们保护环境、回归自然的需求[1-2],其有望代替粘胶纤维。目前,国内Lyocell纤维产业化项目取得重大突破,中国纺织院绿色纤维股份公司的首条年产3万吨Lyocell纤维生产线成功运行[3];江苏金荣泰6万吨Lyocell短纤维以及湖北金环新材料股份有限公司4万吨Lyocell短纤维项目一期也即将投产。此外,国内在Lyocell纤维长丝方面也取得长足进步,2017年1月正式投入量产“龙赛尔”长丝,开创了我国莱赛尔纤维的新品类[4],弥补了行业中长期存在的高档纤维供给不足的难题[5]。
但是,在Lyocell纤维工业化生产中,存在纺丝原液不稳定、溶剂回收率低等技术问题,一般是通过添加稳定剂来改善这种情况。目前,没食子酸丙酯是最常用提高纤维素/NMMO/水三元体系稳定性的试剂;实践证明,PG的稳定性效果非常理想,但PG的加入会使Lyocell纤维纺丝原液变黄,最终影响纤维产品的颜色以及后续染色时的着色性等[6-8]。因此,本文按照文献[9]尝试合成新型稳定剂2,4,5,7,8-五甲基-4-氢-1,3-苯并二噁英-6-醇,并将其运用在Lyocell纤维纺丝原液中,以期可以达到在稳定性与颜色方面均良好的效果,最终取代或部分取代PG。
1 试 验
1.1 试剂
三甲基氢醌,分析纯,Sigma-Aldrich Trading;乙醛,分析纯,Sigma-Aldrich Trading;冰醋酸,分析纯,上海凌峰化学试剂有限公司;浓盐酸,化学纯,太仓沪试试剂有限公司;乙醇,化学纯,国药集团化学试剂有限公司;丙酮,分析纯,国药集团化学试剂有限公司;氘代氯仿,分析纯,Sigma-Aldrich Trading;N-甲基吗啉氧化物,印度Amines & plasticizersLtd公司,50%;没食子酸丙酯,化学纯,国药集团化学试剂有限公司;浆粕(DP=820、α-纤维素含量97.9%),美国Rayonier Inc公司。
1.2 仪器
傅里叶变换红外光谱仪,Nicolet 8700型,美国热电公司;核磁共振波谱仪,AVANCE600型,瑞士Bruker公司;拉曼光谱仪,Invia-Reflex型,英国雷尼绍公司;透射偏光显微镜,XP-550/500C型,上海蔡康光学仪器有限公司;气相色谱质谱联用仪,QP2010ultra型,日本岛津公司;流变分析仪,RSl50L型,德国HAKKE公司;紫外光谱分析仪,Biomate 3S型,美国Thermo Scientific公司。
1.3 PBD的合成及纯化
PBD合成的反应方程式如图1所示。由于TMHQ在空气中容易被氧化,合成反应须在氮气气氛中进行,鉴于乙醛的沸点低,因此,氮气置换空气后即停止通入防止将乙醛带走。具体过程如下:将100 mmol三甲基氢醌与200 mL冰醋酸的悬浮液在氮气气氛中冷却至0℃,缓慢加入20 mL浓HCl(催化剂)继续冷却至 0℃后,停止通入氮气并将反应体系密封,采用分液漏斗加入200 mmol 乙醛,搅拌3 h后停止反应。将反应混合物倒入1 500 mL冰水中,搅拌洗涤1 h后过滤,并依次用50 mL乙酸水溶液、250 mL水洗涤,最后将其在乙醇水溶液(体积比1∶1)中重结晶,干燥后最终得到产物PBD。

图1 PBD的合成方程式
1.4 合成产物的结构表征
气相色谱质谱联用分析:将PBD溶解在丙酮中配成浓度为2 mg/mL的溶液,采用顶空进样的方式测定样品的保留时间,确定样品的组份。
傅里叶变换红外光谱分析:取适量干燥的PBD采用KBr压片法制样,扫描波数范围为400~4 000 cm-1,分辨率为4 cm-1,扫描次数为32次。
核磁共振能谱分析:将PBD溶解在ClCD3中配制成浓度为2 mg/mL的溶液,对其进行核磁共振氢谱、核磁共振碳谱、DEPT135度谱和DEPT90度谱测试;测试条件:采样次数1 024次,采样时间0.90 s,脉冲宽度4 μs,延迟时间2 s,频率600 MHz。
拉曼光谱分析:测试所用激发波长为532 nm。
1.5 纺丝原液以及浸出液的制备
将浆粕用粉碎机粉碎后,放置在50℃的烘箱中4 h,除水使其达到恒重,以减小实验误差;在50%的NMMO溶剂中加入质量分数0.1%的稳定剂后将其浓缩至87%,制备质量浓度为5%的纺丝原液。溶解釜设置温度为90℃,搅拌速度为120 r/min,搅拌时间为2 h。最后,将制得的纺丝原液取合适的质量浸泡在其质量150倍的去离子水中24 h,制得纺丝原液浸出液。
1.6 纺丝原液以及浸出液的分析
流变分析:在稳态剪切模式下对纺丝液进行流变性能测试,剪切速率为0.01~1 000 s-1,测试温度为90℃,锥板直径为35 mm,测试间隙为0.056 mm。
紫外光谱分析:取适量的浸出液进行紫外光谱测试,去离子水作为参比液。
2 结果与讨论
2.1 PBD合成过程中影响因素的探讨
2.1.1 搅拌方式对PBD合成的影响
考虑到乙醛的沸点较低以及反应物需要分批加入,在实验中设置分液漏斗依次加入物料。另外,实验过程中采用低温冷却循环装置,设置温度为0℃;低温冷却液为10%的乙二醇水溶液,防止水在0℃时结冰影响循环效果。随着产物的生成,反应体系的浓度越来越大,低温循环装置自带的转子已经不足以将反应物搅拌均匀,因此本文中采取机械搅拌的方式进行合成反应,此时的加料顺序为乙醛在催化剂盐酸之后加入,最终合成产物如图2所示,为针状晶体。

图2 合成产物
2.1.2 加料顺序对PBD合成的影响
(1) 催化剂在乙醛之后加入
将三甲基氢醌与乙酸悬浮液冷却至0℃后加入乙醛,之后再加入催化剂盐酸,在机械搅拌的方式下反应3 h后将产物干燥后如图3所示。

图3 先加入乙醛后的合成产物
观察图3中并没有明显的文献中所描述的晶状物质。为了进一步验证产物的存在状态,将产物与三甲基氢醌进行气相色谱测试如图4和图5所示。从图4和图5中可以看到产物是三甲基氢醌与PBD的混合物,且大部分都是三甲基氢醌。由此说明,在保证实验环境密闭的情况下,后加入催化剂盐酸时产率较低,可能的原因是盐酸在乙醛之后加入会放出大量的热,乙醛损失掉一部分。
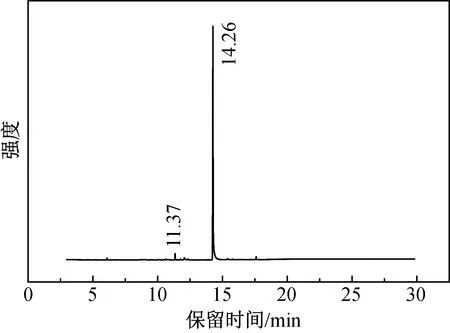
图4 三甲基氢醌的GC图谱
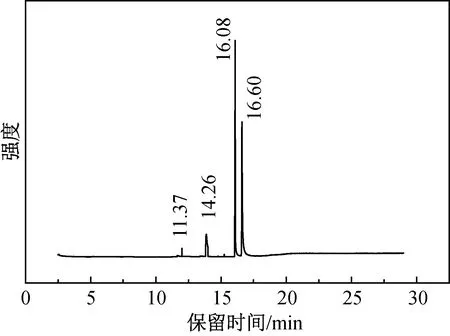
图5 合成产物的GC图谱
(2) 催化剂在乙醛之前加入
在机械搅拌下将盐酸催化剂在乙醛之前加入的实验结果如图2所示。由此可知,在此合成反应中催化剂的加入顺序直接影响到实验的产率,催化剂盐酸应在乙醛之前加入。
2.2 PBD的形貌、纯度、结构分析
2.2.1 PBD的形貌
图6是将PBD在偏光显微镜下观察所得,可以看到PBD是针状晶体。
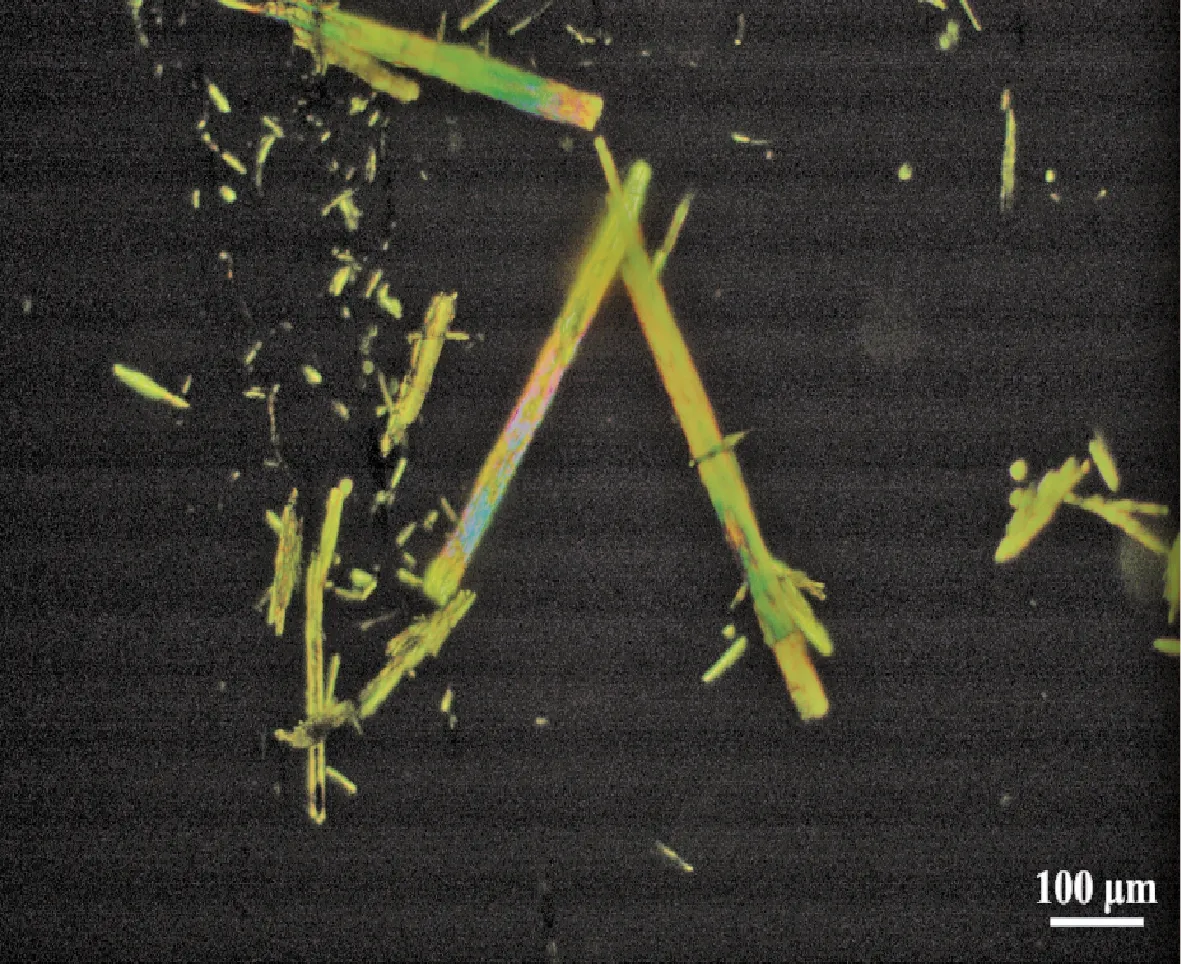
图6 PBD的偏光显微镜照片
2.2.2 PBD的纯度分析
采用气相色谱测试目标产物的纯度。合成反应中可能存在的固体物质为三甲基氢醌和PBD两种物质,图7和图8分别是产物重结晶前后的GC图谱。因此从图7和图8可知,产物在重结晶前明显时还残存反应物三甲基氢醌,对产物进行重结晶提纯后,产物的纯度基本可认为是100%的;另外可知,三甲基氢醌在气相色谱中的保留时间为11.39 min 和14.26 min左右;PBD的保留时间为16.29 min和16.81min左右。
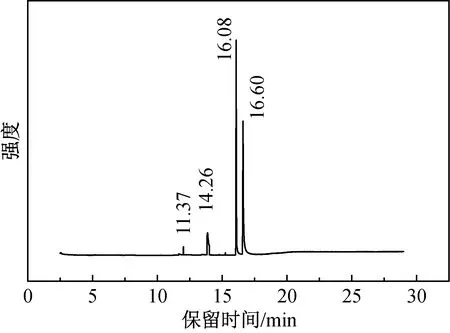
图7 重结晶前产物的GC图谱
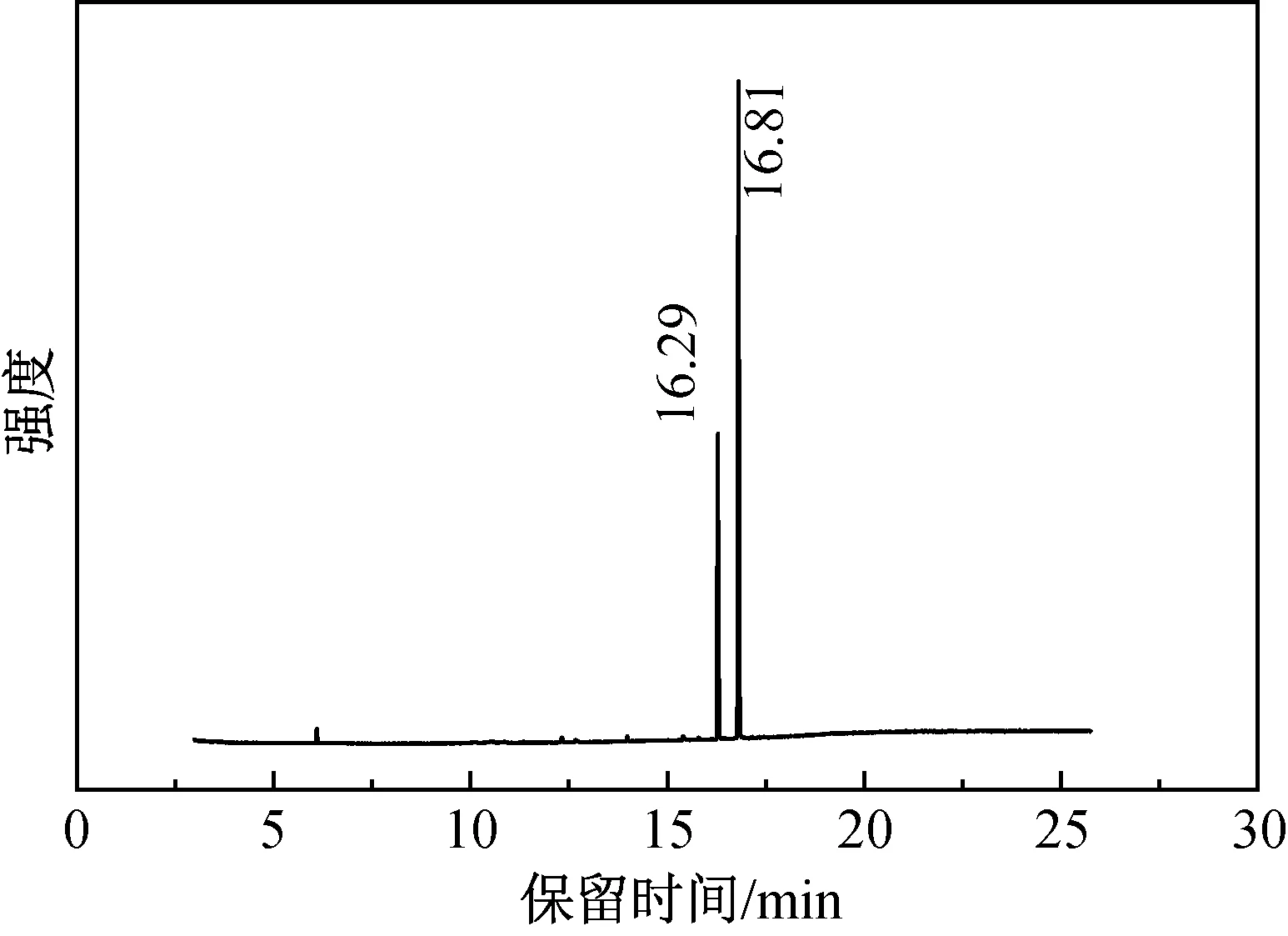
图8 重结晶后产物的GC图谱
2.2.3 PBD的红外、拉曼光谱分析
将PBD提纯之后,进一步确定其结构特征,采用溴化钾压片方式对PBD进行红外测试,其结果如图9所示。从图中我们可以看出,实验产物在3 650~3 200 cm-1官能团区3 410 cm-1处有宽且强的酚羟基吸收峰,这是由于羟基在形成氢键缔合后,O--H+键拉长,电偶极矩增大,因此在3 650~3 200 cm-1表现为一个强而宽的峰;在2 990 cm-1处有较弱的C-H收缩振动峰;而在波数为1 371 cm-1处出现甲基单峰,可以判断分子中有甲基;在波数1 600~1 430cm-1间于1 460 cm-1处出现苯环骨架振动吸收峰,分子中有苯环;在857、797、721和642 cm-1处出现苯环上C-H面外弯曲振动吸收峰,并且在波数895 cm-1出现五取代苯环强吸收峰;并且,在X-Y伸缩振动区1 096 cm-1处出现C-O键伸缩振动吸收峰以及1 257 cm-1处出现C-O-C键收缩振动吸收峰,可见产物的分子结构中还可能有C-O或C-O-C键。拉曼光谱是对红外光谱的补充,我们对PBD进行两次拉曼光谱扫描,结果如图10所示,其在2 990 cm-1处有强吸收峰对应C-H收缩振动峰。由此可知,PBD的主要结构框架为具有酚羟基和其他五取代的芳香结构,另外分子中还含有C-O结构。

图9 PBD的红外图谱
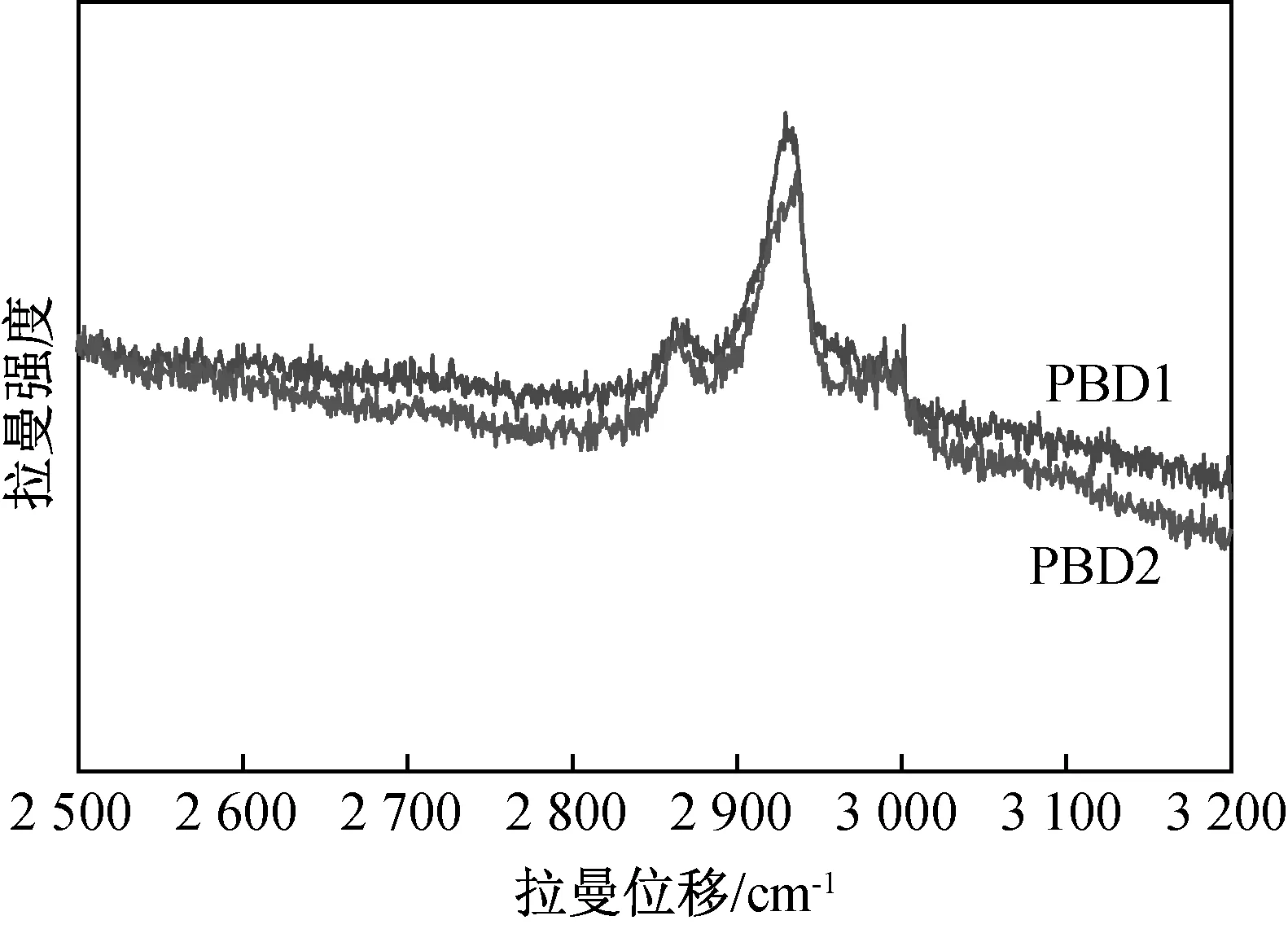
图10 PBD的拉曼图谱
2.2.4 PBD核磁共振分析
通过上述红外与拉曼光谱对于PBD结构的初步判定,对PBD再进行核磁共振测试。将PBD溶在氘代溶剂氯仿(CDCl3)中,对其进行1H核磁共振谱和13C核磁共振谱、DEPT135度谱、DEPT90度谱测试。其中,DEPT谱为无畸变极化转移增强,是一种13C核磁共振谱中的一种检测技术,主要用于区分13C谱图中的伯碳、仲碳、叔碳和季碳(不出峰);135度的DEPT谱图:CH、CH3的峰向上(即信号为正),CH2为倒峰(即信号为负),90度的DEPT谱图:只能看到CH向上的峰。因此,由1HNMR谱图11可知,δ(化学位移)1.48,1.55对应的基团为CH3、δ 2.12,2.15,2.18对应的基团为ArC-CH3、δ 4.36对应的是OH、δ 4.95,5.02,5.23,5.33对应的基团为CH;由核磁碳谱图11和图12可知,δ 11.7,12.2,12.7,20.8,21.1对应的为伯碳CH3、δ 69.1,71.2,89.6,95.6对应的为叔碳CH、δ116.4,121.3,122.2,122.9,145.8,146.2对应的为季碳。此结论与文献[9]中的结论也是一致。

图11 PBD的1H谱图
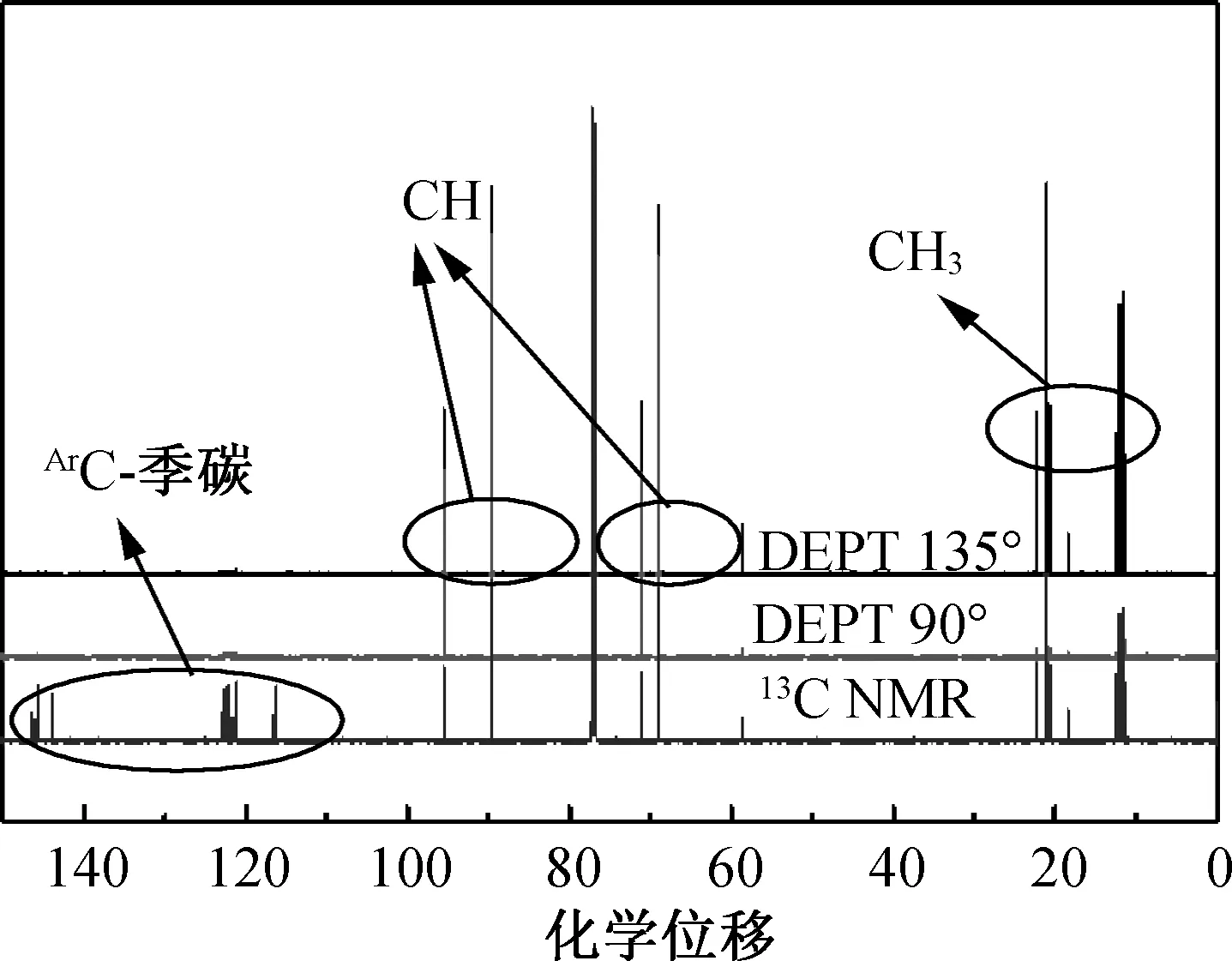
图12 PBD的13C谱图
2.3 PBD对纤维素/NMMO/水溶液的稳定性分析
2.3.1 凝固浴的紫外吸光度分析
对于发色物质的检测,紫外是目前为止最突出最广泛使用的技术[10]。图13是添加不同稳定剂后,紫外分光光谱仪测得的纺丝原液浸出液的吸光度值。
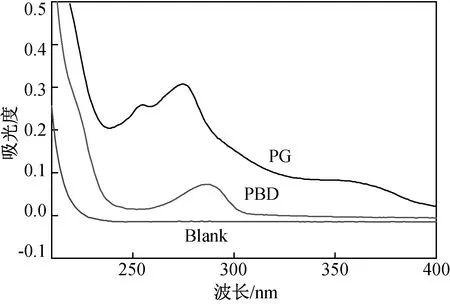
图13 添加不同稳定剂后纺丝液凝固浴的吸光度值
从图13中可以看出添加PBD、PG的纺丝原液浸出液均出现了吸收峰,表明添加稳定剂之后都会有发色团的出现,紫外光谱的生色基是碳碳共轭结构、含有杂原子的共轭结构、能进行n→π*跃迁的基团、能进行n→σ*跃迁并在近紫外区能吸收的原子或基团,显然在250~300 nm之间出现的生色基吸收峰对应的是带有苯环的物质。进一步比较图9可知,添加稳定剂PG的纺丝原液浸出液产生的生色基团的量明显大于PBD,说明在抑制颜色产生方面PBD的稳定作用是优于PG的。
2.3.2 纺丝原液的流变学分析
将添加不同稳定剂的纺丝原液在90℃下进行稳态流变学测试,如图14所示。
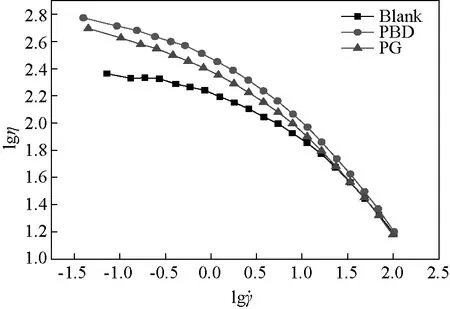
图14 添加不同稳定剂后纺丝原液的黏度变化
从图中可以看出纺丝液的黏度总会随着剪切速率的增大而减小;并且添加稳定剂PBD的纺丝原液黏度要高于添加稳定剂PG的纺丝原液。稳定剂的作用机理为稳定剂分子中的羟基与体系中的自由基进行反应[8],从而降低了自由基攻击纤维素分子链的几率,抑制纤维素降解,使所配制的纺丝原液黏度下降不多,有利于后续纺丝的均匀性以及纤维的力学性能等。基于上述讨论可以得出,无论在纺丝原液浸出液的颜色以及抑制纺丝原液降解方面,稳定剂PBD的效果均优于PG。
2.3.3 稳定剂的稳定机理分析
酚类稳定剂的作用效果基本都是羟基的作用,反应体系中自由基与稳定剂中的羟基发生反应,减少攻击纤维素分子链,从而抑制纤维素降解。具体而言PG的作用机理如图15所示。PG与纤维素/NMMO/水体系中的自由基结合先形成鞣花酸,而鞣花酸会继续与自由基反应,氧化为稳定的双邻醌,而醌类是使纺丝液颜色产生变化的一个主要原因,也就是说PG起到了消灭自由基的作用,理论上一个PG分子能消灭3个自由基。PBD的作用机理如图16所示,与PG作用机理相似,PBD也是起到了消灭自由基的作用,一个PBD分子最终会中和4个自由基。但是,PBD在形成相对稳定的苯氧基自由基后,C-O键断裂发生分子重排又形成了羟基,进而增强了稳定效果。根据图16可知反应最终也是生成了醌类,但是颜色的加深程度明显低于PG。因此,从整体上来讲PBD的稳定效果优于PG。
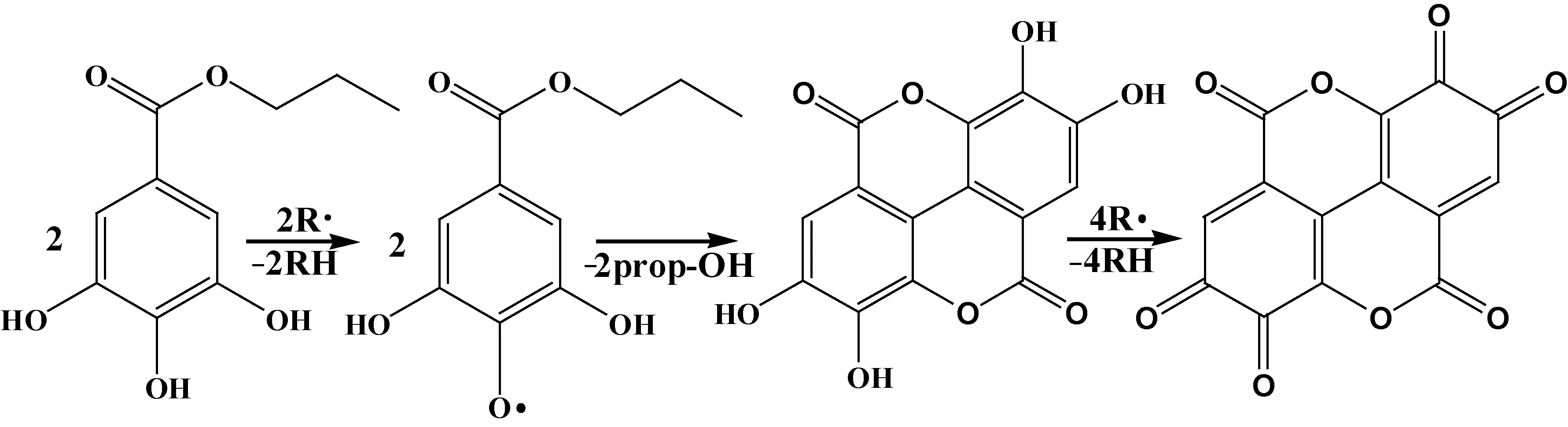
图15 PG的稳定机理
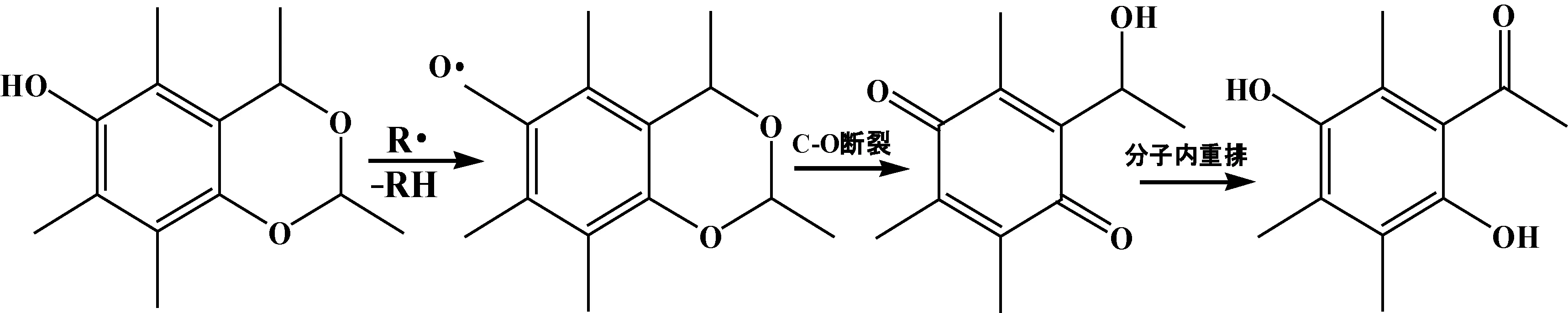
图16 PBD的稳定机理
3 结 论
采用三甲基氢醌与乙醛在低温下反应成功地合成了稳定剂PBD,其为针状晶体,搅拌方式以及加料顺序对PBD的合成具有影响;将PBD应用到纤维素/NMMO/水溶液体系中,发现PBD对纤维素/NMMO/水溶液的颜色以及稳定性均优于PG。
猜你喜欢
浙江化工(2022年8期)2022-09-05
能源化工(2021年6期)2021-12-30
纺织科学研究(2021年1期)2021-12-03
石油沥青(2021年4期)2021-10-14
石油沥青(2021年6期)2021-02-10
纺织科学研究(2020年10期)2020-11-09
名城绘(2020年1期)2020-10-21
纺织报告(2020年4期)2020-08-14
休闲读品·天下(2020年4期)2020-02-04
名城绘(2020年10期)2020-01-03
