
碱土金属吸附二维α1硼烯结构和性质研究
2020-05-13 08:45韩健伟边伟樾罗有华
原子与分子物理学报 2020年4期
韩健伟, 边伟樾, 刘 锦, 徐 豪, 张 孟, 王 潇, 罗有华
(华东理工大学,上海 200237)
1引 言
随着2004年Novoselov等人微机械剥离制备单层石墨烯(graphene)的成功,以石墨烯为代表的新型二维晶体材料因其独特的几何结构和物理化学性质迅速成为了国内外的研究热点[1].硼元素的电子结构为1s22s22p1,同时具有金属和非金属的双重属性,表现出独特的成键方式.2007年,Tang等人通过理论计算发现了一种由三角形晶格和六角蜂窝状孔洞两者混合而成的新型α-sheet硼单层结构[2,3].2012年曾晓成课题组进一步通过CALYPSO程序计算提出了多种形式的二维硼原子层,并且预测了能量上稳定的α1和带皱褶的α'型硼烯结构[4,5].随后,国内外不同研究小组分别计算模拟了在Au(111)、Ag(111)以及 MgB2等衬底生长出单层硼烯的可能性,为实验上的成功制备硼烯指出了方案[6-8].最近中美两国科学家同时在金属衬底上成功制备出了单层硼墨烯, 对将来可能的基于硼烯的潜在应用提供了诱人的前景[9,10].
在二维硼材料领域实验和理论已经开展了很多有价值的工作,目前这些研究主要集中在硼平面材料的结构预测与分析、不同结构的稳定性以及电子性质等.而对二维硼烯的吸附特性以及金属原子吸附硼烯后体系的稳定性、结构变化和电子结构等问题的研究鲜有报道.另一方面,通过吸附和掺杂外来原子等化学修饰方法是提高纳米材料结构稳定性以及改变材料物理化学性质的重要手段[11-15].硼原子具有复杂的成键机制和多配位能力,同时由于缺电子特性,使得硼原子易于与具有给电子特性的金属原子发生相互作用,形成金属硼化物,因此可以通过吸附不同金属的种类来实现调控硼烯的性质.之后谭心团队细致地分析了碱金属吸附在石墨烯和磷烯表面的吸附性质和迁移行为,发现碱金属在两种二维纳米材料表面的最稳定吸附位是空位,并且随着原子序数的增大体系的迁移行为越明显[16-17].2014年,Banerjee等人使用密度泛函方法结合从头算分子动力学模拟研究了α1硼烯吸附锂原子的情况,发现锂原子吸附在硼烯六角空位正上方在能量上是最稳定的,并进一步讨论了硼烯作为锂离子电池阳极材料的可行性[18].;随后Zheng 等人利用密度泛函理论计算研究了碱金属锂、钠和钾原子吸附于α硼烯后体系的稳定性和功函数变化[19,20],目前已有的文献集中在碱金属吸附硼烯的研究情况.碱土金属最外电子层上有两个s价电子,容易失去电子,因而可以和缺电子的硼形成稳定的碱土金属硼化物.据我们所知,目前还没有相关的碱土金属原子或团簇吸附硼烯的文献报道.
本论文工作采用第一性原理对新型α1硼烯吸附碱土金属原子(铍、镁和钙)及其二聚体团簇后体系的几何结构、最低吸附点位、稳定性以及相互作用进行系统地研究.探索碱土金属原子及其二聚体对二维硼烯结构和稳定性的影响,找到最佳吸附位置,并进一步研究复合体的吸附能、电荷转移、态密度等性质,揭示硼烯中硼原子与外来碱土金属原子的成键模式,为今后新型二维硼烯材料的性能调控提供可靠的理论依据.
2计算模型和方法
本文采用基于第一性原理的密度泛函理论(DFT)方法对铍、镁、钙三种碱土金属原子及其二聚体团簇在硼烯表面的吸附性质进行研究.所使用的硼烯为理论上最稳定α1硼烯并且采用(3×3)的超晶胞进行模拟计算,计算软件为Materials Studio 中的CASTEP模块,α1硼烯的真空层设置为15 Å,截断能设为190 eV,布里渊区的K点在Monhkorst-Pack方法计算下选为2×2×1,能量的收敛值为2.0e-5 eV/atom,最大的应力收敛值为0.05 eV/Å,最大压强收敛值为0.1 GPa,最大位移收敛值为0.002 Å,最大迭代次数为200次,并且使用广义梯度近似GGA-PBE泛函对体系进行几何结构优化,能量以及其它性质的计算.经研究发现碱土金属原子吸附于硼烯的位置主要有以下四种:空心位、顶位、桥位以及三角位,并且硼烯中具有一定的对称性,一个充满的六边形硼烯中拥有7个硼原子,考虑等价位之后有3中不同的硼原子B1、B5、B13[5,6],最后我们还列出了在吸附过程中遇到的两种特殊吸附位Near B13以及Near Hollow,如图1所示,为了比较不同体系的稳定性,本文定义了硼烯结构吸附能:Eb=EB+EAM-ET,其中,Eb为总吸附能,EB为纯的硼烯的能量;EAM为孤立碱土金属原子的能量;ET为硼烯吸附碱土金属后复合体的总能量.吸附能若是负值表明这个体系时无法共存的,而吸附能的值为正的,则表明有相互作用.计算所得吸附能值越大,表明碱土金属原子与硼烯结合作用越强;反之,表明二者的结合作用越弱.
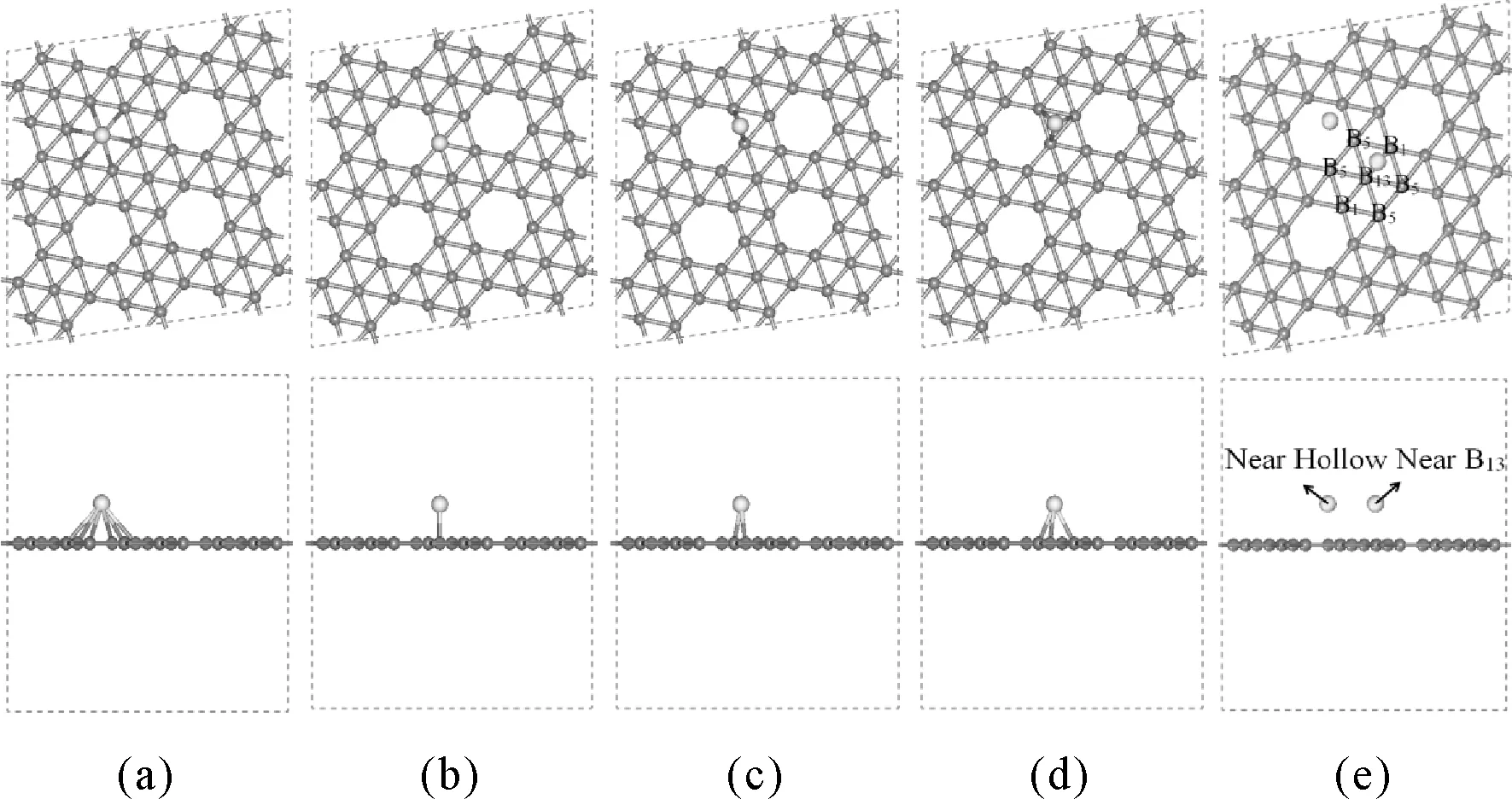
图1 α1硼烯表面五个高对称吸附位的俯视图和侧视图. (a)空心位Hollow,(b)顶位Top,(c)桥位Bridge,(d)三角位Triangle,(e)Near B13以及Near HollowFig. 1 Top view and side view offive high symmetry adsorption sites of borophene. (a)Hollow,(b)Top,(c)Bridge,(d)Triangle,(e)Near B13 and Near Hollow
3结果和讨论
3.1单个铍、镁、钙原子在α1硼烯表面的吸附性质
由于α1硼烯是通过硼原子以三角形和六边形混合的组合方式排列的,因此整个平面的对称度是不如石墨烯的(所有的碳原子都是等价的),本工作选取拥有63个硼原子的(3×3)的超晶胞,其中有三种不同的硼原子,导致同一类高对称吸附位就存在多个不同的吸附位置,为了便于比较,经过计算我们找到了每一类吸附位中吸附能最高的或吸附特征较为特殊的几种,并就对这几种不同的碱土金属的不同吸附位中吸附能最大的一种进行比较,得到的碱土金属—硼烯体系的能量和吸附能见表1和最稳态图2.
从表1和图2可以看出,在碱土金属—硼烯的体系中,三种不同的碱土金属Be、Mg和Ca无论在何种位置进行吸附体系的吸附能总是正的,表明碱土金属与α1硼烯确凿发生了相互作用,应当存在电荷之间的转移或移动从而发生化学吸附.分析数据可以看到三种碱土金属吸附在α1硼烯表面时的特征是具有较大区别的.对于铍原子来说,当吸附的位置选择除了Top(B13)这个位置,无论是其它的顶位,抑或是桥位、三角位、空心位,最终稳定的位置都是靠近原吸附位最近的一个空位,而在Top(B13)这个位置铍原子会最终稳定在最初吸附的位置,可见铍原子吸附在α1硼烯只有两个稳定的位置,并且从吸附能可以看到当铍原子稳定在空位时是最稳定的,Top(B13)只是一个亚稳态,两者总能量相差2.025 eV左右.对于镁原子来说吸附的位置全部与最初的试探位是相一致的,可见上文所提到的所有的可能的吸附位,对于镁原子来说都是可以稳定的,并且在空位吸附时镁原子具有最大的吸附能.而对于钙原子来说情况稍显复杂,总结一下可以知晓,钙原子吸附在α1硼烯上大抵有三个较为稳定的位置,Near B13、Near Hollow以及空心位,具体吸附的位置与最初的试探位置有关,试探位靠近哪个位置,则最终就会吸附在那个位置,并且前两个位置的吸附能差距并不是很大,Near B13与Near Hollow体系总能量相差0.03 eV,而单独稳定在Hollow位时的吸附能是最小的.
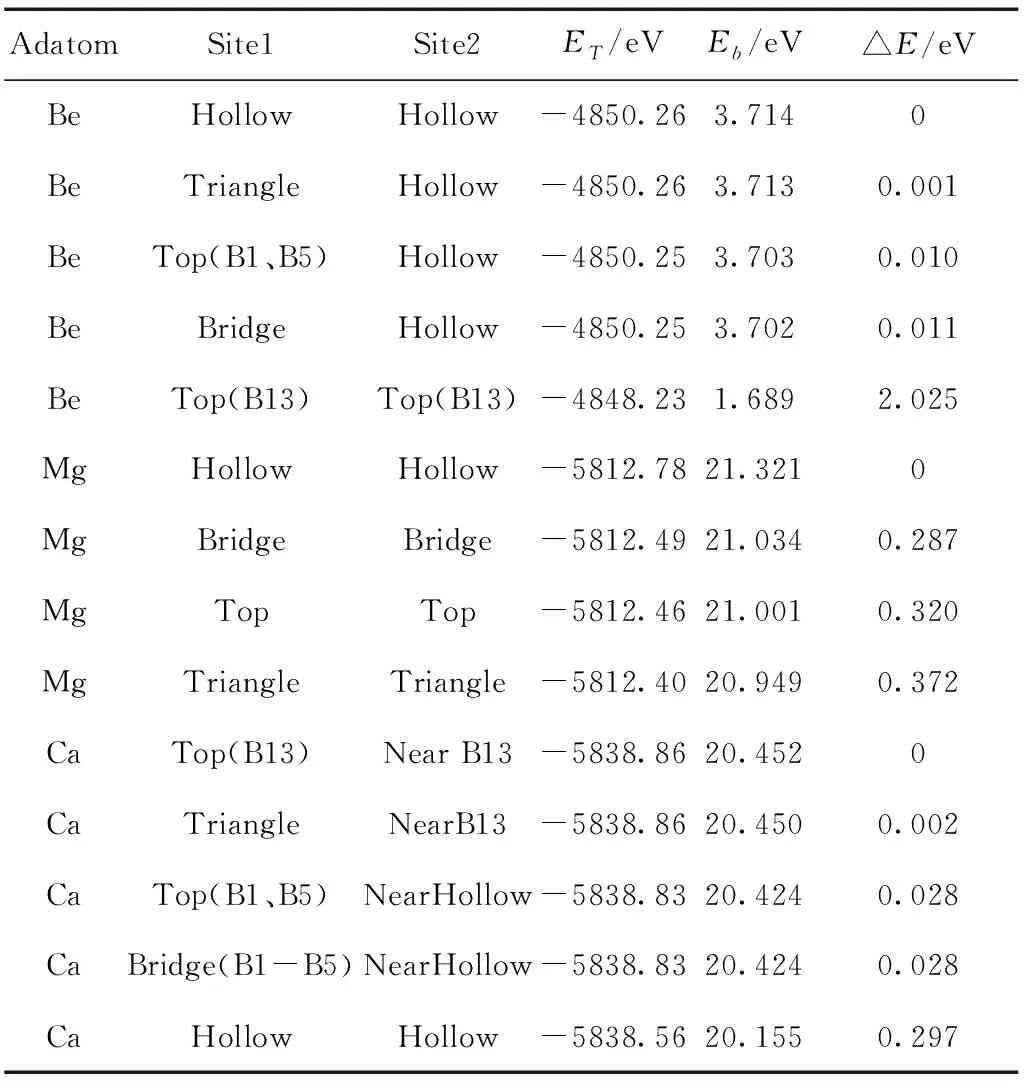
表1 碱土金属原子在α1硼烯表面不同吸附位的吸附能.其中,Site1为优化前初始试探位,Site2为优化后最终吸附位,ET为体系总能量,Eb为吸附能,△E为与最稳态的相对能量差

图2 铍、镁、钙原子吸附在α1硼烯上最稳态的俯视图和侧视图,(a)、(b)和(c)分别对应铍、镁、钙原子Fig. 2 The most stable top and side views of beryllium, magnesium and calcium atoms adsorbed on α1 borophene. (a), (b), and (c) correspond to beryllium, magnesium and calcium atoms, respectively.
值得注意的是在文献19、20中,Li 原子处于α1硼烯不同位置的预设结构优化后均指向六边形孔(Hollow位)中心上方的位置,与我们工作中Be和Mg的结果是一致的. 但Ca原子的最低能量吸附位置发生了改变,根据我们的计算,Near B13(非正上方)是最稳定的吸附位置.
比较三种碱土金属吸附在α1硼烯表面的吸附能可以看到,镁原子与α1硼烯的吸附能是最大的,钙原子次之,而铍原子虽然与α1硼烯存在相互作用,但吸附能约为3.7 eV,明显小于Mg和Ca碱土金属原子吸附的情况,但高于文献17、18报道的单个Li原子的吸附能2.33 eV.另外我们测量了碱土金属吸附在α1硼烯表面的距离,发现铍原子与硼烯表面的距离为0.739 Å,镁原子为1.991 Å,而钙原子为2.532 Å,铍原子与α1硼烯表面的距离过近,对α1硼烯表面的破坏程度是最大的,因此吸附能最小,钙原子虽然具有较大的原子半径,但它与硼烯表面的距离比镁原子要大,这可能是钙原子的吸附能略小于镁原子的原因.综上α1硼烯具有较强的储镁以及储钙的功能,由于铍原子特殊的吸附位,α1硼烯在储铍方面也存在应用的空间,潜力较大.
3.2单个碱土金属吸附在最稳处时的性质研究
在找到碱土金属原子吸附α1硼烯的能量最低的吸附位置基础上,通过计算所研究体系的Mulliken布局分析以及电子态密度图来进一步研究α1硼烯吸附碱土金属原子后体系的电子结构性质,分析其相互作用. 从Mulliken布局分析可以得到铍原子,镁原子和钙原子在吸附后分别失去了1.35e,1.39e和1.30e电量的电荷,这些电子主要来自于最外层s轨道.对于铍原子来说,铍原子所失去的电子大都转移到了六边形孔洞上的4个B5原子和两个B1原子上,4个B5原子各自获得了0.21e,而两个B1原子分别获得了0.25e,可见铍原子与硼烯中六边形孔洞硼环的6个硼原子之间具有很强的等量电荷交换,铍与整个硼烯体系的作用几乎局限于六边形孔洞附近.对于镁原子,它所失去的电子则分散的相对较广,其中六边形孔洞上的4个B5原子各自获得了0.23e,而两个B1原子各自获得了0.11e,并且镁原子的部分电子转移到了距离六边形孔洞较远的硼原子上,可见相对于铍原子,镁原子与整个α1硼烯表面的作用要更充分.钙原子的稳定位不同于铍、镁原子,在吸附过程中转移的电子大都转移到了被充满的六边形上,6个硼原子各自平均获得0.12e,并且同镁原子一样其所转移的电子分散在整个α1硼烯中,因此与硼烯表面的作用范围较大,而与镁原子不同的是,B13在吸附过程中同样也失去了0.06e的电子,因此B13与钙原子存在一定的电子之间排斥力,这也是钙原子吸附能小于镁原子的另一个原因.三种碱土金属的共同点在于,距离稳定位较远的部分硼原子同样也失去了较少的电子,并且α1硼烯在六边形结构附近是极易得到电子的,且平面整体呈现离域性.
图3为三种不同的碱土金属吸附在α1硼烯最稳态上的一系列电子态密度(PDOS)图,虚线代表费米能级.从(a)中可以看到主要是体系的s和p轨道都穿过了费米能级,说明吸附后的α1硼烯没有带隙,和纯的硼烯一样呈现金属特性.从(b)、(c)、(d)图中可以看到碱土金属都是外层s轨道的电子转移到了硼烯上,并与其产生了相互作用.铍原子吸附在α1硼烯后的电子态密度产生了很大的变化,当铍原子与硼原子相互作用时,其s轨道上的电子被部分激发,来到了原本空着的三个高能级p轨道,其s态和p态的电子同α1硼烯上硼原子上的电子相互作用产生了s-p以及p-p共价键. 镁原子由于吸附在α1硼烯表面上的距离大于铍原子,其s轨道上的电子只有一小部分被激发来到了p轨道同硼原子产生了共价键.钙原子吸附在α1硼烯表面上的距离是最大的(2.532 Å),其原本占据在s轨道的电子绝大部分离开了原本的轨道了,来到了α1硼烯上,因此钙原子与硼烯主要以离子键为主,这也解释了为什么钙的吸附能小于镁.

图3 碱土金属在α1硼烯最稳态上的PDOS图.(a)为α1硼烯以及三种碱土金属吸附在α1硼烯上总的PDOS图,(b)、(c)、(d)分别为孤立的铍、镁、钙原子的PDOS图以及吸附在α1硼烯上后铍、镁、钙原子的PDOS图Fig. 3 The PDOS diagrams of the alkaline earth metal in the most stable state of α1 borophene. (a) is the total PDOS diagram of α1 borophene and three alkaline earth metals adsorbed on α1 borophene, and (b),(c)and (d) are PDOS diagrams of free magnesium and calcium atoms and PDOS diagrams of beryllium, magnesium and calcium atoms adsorbed on α1 borophene, respectively.
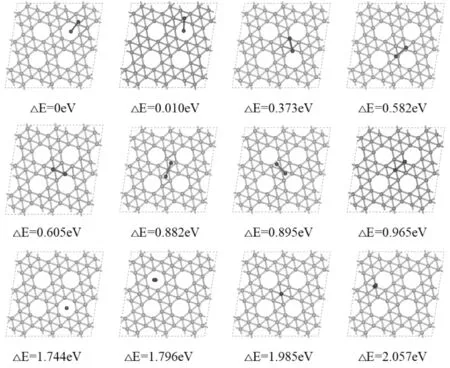
图4 铍二聚体团簇吸附在α1硼烯上的前12种最稳构型及其与最稳定构型的相对能量差Fig.4 The first 12 most stable configurations of Be clusters adsorbed on α1 borophene and their relative energy differences with the most stable configuration
3.3碱土金属双原子团簇在α1硼烯表面的吸附性质
为了进一步考察多个碱土金属原子吸附在α1硼烯上的几何结构,稳定性以及对应的性质,我们同时对碱土金属双原子二聚体团簇在α1硼烯表面的吸附进行进一步的探索.显然,二聚体团簇吸附硼烯的可能初始吸附位置要比单原子复杂的多,我们考虑了众多可能的吸附位置和吸附方式,并进行了结构优化.找到了碱土金属二聚体团簇吸附在α1硼烯上几种稳定构型,并选取了吸附能最大的前十二种构型整理如图4、5、6所示,排布顺序按照吸附能由大到小排列.
如图4所示当铍团簇的初始试探位位于空的六边形孔洞附近时,团簇首先会缩短键长,然后一个原子会慢慢来到六边形空洞的中心,另一个来到B1或B5,这两个都是稳定点,B5比B1相对更稳定.硼烯整体对铍原子具有较强的吸引力,原子距离硼烯表面较近,其中一个铍原子对B5或B1形成一定程度的挤压,造成B5或B1略微下移.而当初始试探位在满的六边形孔洞周围时,吸附过程中铍团簇首先也会缩短键长,通过挤压硼烯表面来达到平衡,但这种情况下,吸附能普遍不是很大.最后我们还研究了铍团簇垂直于硼烯表面时的吸附情况,发现吸附能普遍不如前两种,其原因推测距离硼烯较远的那个铍原子与硼烯基底几乎没有相互作用了,具体吸附情况如图4所示.
图5是镁团簇吸附在α1型硼烯上的情况,我们发现吸附能最大的地方同样也是其中一个镁原子在Hollow位,而另一个镁原子位于距离下一个Hollow位较近的地方或者其它吸附位(Top,Bridge,Triangle),最低能量构型中两个镁原子倾向于各自吸附于单个镁原子的最稳定吸附位置(Hollow位).当初始试探位位于充满的六边形孔洞附近时,镁团簇的吸附能就普遍不是很高.最后我们同样也尝试了垂直于硼烯表面的情况,发现吸附能是最低的,这些与铍团簇的吸附情况是大致一样的.而与铍团簇不同的是,镁团簇的亚稳态点明显多于铍团簇,在很多位置都能稳定(如图5所示).此外,镁团簇吸附在α1硼烯上时,最稳定的几种吸附构型大多倾向于一个镁原子吸附在Hollow位,即使改变初始构型的吸附点,优化后一个镁原子始终稳定在Hollow位,从中看出α1硼烯的Hollow位对单个镁原子具有很强的吸引作用,这个和前面单个镁原子在Hollow位具有较大的吸附能(21.321 eV)相吻合.

图5 镁二聚体团簇吸附在α1硼烯上的前12种稳定构型及其与最稳定构型的相对能量差Fig.5 The first 12 most stable configurations of Mg clusters adsorbed on α1 borophene and their relative energy differences with the most stable configuration
图6为钙团簇的吸附情况,发现当一个钙原子吸附在靠近B13附近,另一个钙原子吸附在六边形孔洞中心的附近时,体系的吸附能是最大的,这分别是单个钙原子吸附时,吸附能最大的两个位置.同镁团簇吸附情况相似的是,当两个钙原子都十分靠近六边形孔洞中心的附近时,体系的吸附能相对是较大的,当钙团簇吸附在被充满的六边形孔洞附近时吸附能次之,垂直情况同其它碱土金属一样是最小的.
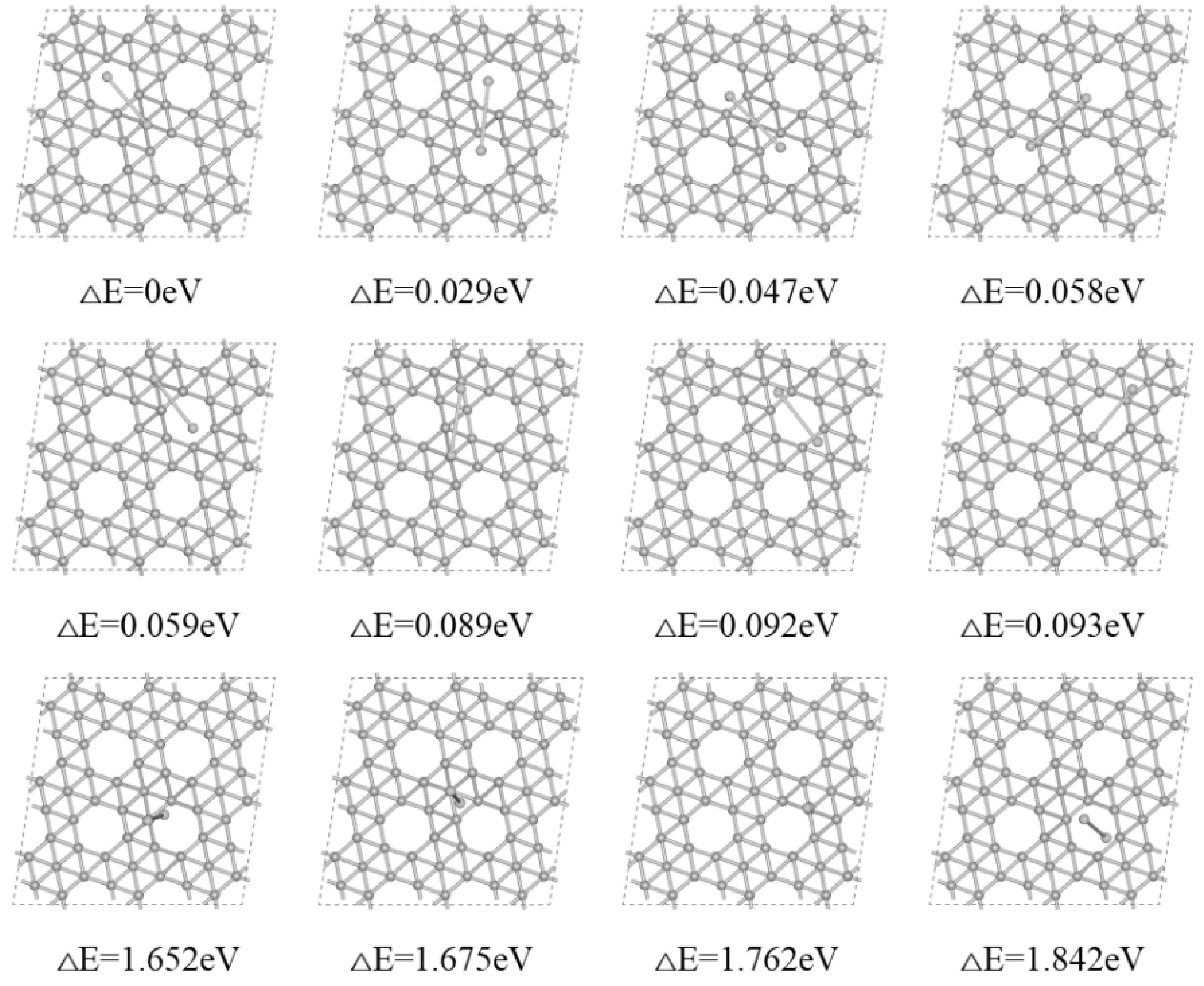
图6 钙二聚体团簇吸附在α1硼烯上的前12种最简构型及其与最稳定构型的相对能量差Fig. 6 The first 12 most stable configurations of Ca clusters adsorbed on α1 borophene and their relative energy differences with the most stable configuration
从表2以及图7中我们不难发现碱土金属双原子团簇吸附在α1硼烯上后,团簇之间的距离会缩短,二聚体团簇的吸附能是稍大于单个碱土金属的吸附能的,然而除了铍原子,团簇的平均吸附能是明显小于单个原子的吸附能,可见α1硼烯更倾向于单个原子的吸附.因此我们还计算了两个碱土金属各自吸附在最稳态时的情况(单个钙原子吸附时Near B13和Near Hollow两个位置的吸附能相差很小,两个碱土金属原子吸附后原子位于Near Hollow的吸附能稍大于Near B13),研究发现当两个碱土金属原子孤立吸附在硼烯后,其吸附能相比于二聚体团簇反而稍大一些,由此我们推测当碱土金属原子吸附在α1硼烯上时,分散的单原子吸附在能量上是较有利的,吸附原子之间不会倾向于聚集成团簇.
4结 论
本文采用基于密度泛函理论的Materials Studio计算软件中的CASTEP模块对碱土金属吸附α1硼烯的最低能量吸附位置、结构稳定性和相互作用进行了系统的计算和研究. 首先对碱土金属(铍、镁和钙)单原子、二聚体团簇、双原子吸附在α1硼烯上最低能量构型进行了鉴定和讨论,在此基础上研究了碱土金属原子与硼烯的相互作用.结果表明:(1)单个铍原子和镁原子吸附在α1硼烯Hollow位是体系吸附能最大的情况,而钙原子在B13附近时体系的吸附能最大.(2)相对于铍原子情况,镁、钙原子吸附在α1硼烯上的吸附能较大,说明α1硼烯在储镁和储钙方面存在应用潜力.(3)通过电荷转移和态密度等性质分析,发现铍原子吸附在最稳态时可以与α1硼烯产生s-p以及p-p共价键,铍与硼原子的共价键最明显.镁原子是离子键和共价键的混合成键方式,而钙原子则主要是离子键形式.(4)对于二聚体吸附情况,双原子铍团簇的最稳态为一个铍原子在Hollow位,另一个在B1附近,镁团簇的最稳态为一个在Hollow位,另一个镁原子在其它的Hollow位附近,钙团簇的最稳态为一个在B13附近,另一个在Hollow的中心附近.(5)二聚体团簇吸附能大于单原子吸附能但小于双原子吸附情况,表明碱土金属原子吸附在α1硼烯上时,吸附原子之间不会倾向于聚集成团簇,在稳定性上反而更倾向于分散的单原子吸附方式.研究结果和前人碱金属吸附的情况进行了对比和讨论,这些研究结果丰富了二维硼烯的体系和结构,并为开发以二维硼烯为基础的功能性纳米材料提供参考价值.

表2 碱土金属双原子团簇吸附在α1硼烯的吸附能,Lc为真空中孤立的双原子团簇间的距离,D为实际优化后两个原子间的距离,Et为总的能量,Eb1为二聚体团簇的吸附能,Eb2为两个碱土金属原子分别吸附在最稳态的吸附能

图7 (a)、(b)、(c)分别为两个铍、镁、钙原子各自吸附在最稳态上的俯视图和侧视图Fig. 7 (a), (b), and (c) are top and side views of the two beryllium, magnesium, and calcium atoms adsorbed on the borophene in the most stable system, respectively
猜你喜欢
大电机技术(2022年3期)2022-08-06
核科学与工程(2021年4期)2022-01-12
小哥白尼(趣味科学)(2021年6期)2021-11-02
电子技术与软件工程(2021年7期)2021-06-16
煤气与热力(2021年4期)2021-06-09
中华戏曲(2020年1期)2020-02-12
收藏界(2019年3期)2019-10-10
童话世界(2018年32期)2018-12-03
学生导报·高中版(2017年23期)2017-09-10
学生导报·初中版(2017年23期)2017-09-10
