
光催化合成吲哚-3-乙酸酯类衍生物反应研究
2020-05-13 05:14张晓斐朱培元姚团利李丹璐
陕西科技大学学报 2020年2期
张晓斐, 朱培元, 姚团利, 李 翔, 解 攀, 李丹璐
(1.陕西科技大学 化学与化工学院, 陕西 西安 710021; 2.陕西科技大学 陕西省轻化工助剂重点实验室, 陕西 西安 710021)
0 引言
吲哚-3-乙酸酯结构是许多天然生物碱和生物医药分子的核心骨架(图1),具有重要的生物活性及较高的合成价值[1-3].如吲哚美辛(Indomethacin),是一种典型抗炎镇痛药物; 阿比米尔(Arbidol),是一种抗病毒药物.

图1 含有吲哚-3-乙酸酯骨架的生物活性分子
目前,许多制备吲哚-3-乙酸酯衍生物的方法被相继报道.其中,邻烯基芳基异腈与有机金属试剂的反应作为构建此类化合物最有效的途径之一[4],受到合成化学家的广泛关注 (图2).Fukuyama课题组以邻烯基芳基异腈为原料,利用Bu3SnH介导的自由基反应合成吲哚-3-乙酸酯衍生物[5],该方法被广泛应用于吲哚类天然产物合成中[6-8].2010年,Mamoru等[9]开发了一种不需Sn,通过Cu(I)催化,合成2-硼酸化的吲哚-3-乙酸酯的方法.这两种策略随后都需要通过钯催化的偶联反应得到2-取代吲哚-3-乙酸酯衍生物.2018年,Heckma等[10]报道了一类Cu(II)催化的环化反应,该方法利用2-烯基异腈与芳基硼酸为原料,可以高效的得到相应的吲哚-3-乙酸酯衍生物.

图2 邻烯基芳基异腈合成吲哚-3-乙酸酯类衍生物
过渡金属催化的脱羧加成反应经过多年的发展,已经能够实现多种类型的转化过程,为构建C-C键,C-杂原子键的构建提供了有效的反应途径[11].但是该类反应仍存在诸多不足,如常需要较高的温度来满足羧酸脱羧所需要的能量,而高温的条件又不利于反应的发展与利用.近年来可见光促进的脱羧加成反应得到了快速发展[12].和传统的过渡金属催化相比,光催化脱羧具有反应条件温和、兼容性好等优点,因此成为化学家研究的热点领域之一.
2013年,Xie等[13]报道了以烷基羧酸为自由基供体,N-甲基-N-芳基甲基丙烯酰胺为自由基受体的光催化脱羧加成反应.2014年,Chu 等[14]利用光催化实现了烷基羧酸与Michael受体的脱羧加成反应.和之前的工作相比,该反应兼容α-氨基酸和α-氧代羧酸,并且不需要在体系中加入额外的氧化剂.2015年,Wang等[15]利用光催化实现了芳基酮酸的脱羧Michael加成反应.该反应适用于醛,酮,腈,以及α、β-不饱和脂等各类Michael受体.虽然光诱导的脱羧加成反应已经被广泛研究,但是以邻烯基芳基异腈为自由基受体,酮酸为自由基供体合成吲哚-3-乙酸酯类衍生物的反应还未见报道.考虑到吲哚-3-乙酸酯衍生物的重要应用价值,开发一种以廉价易得的酮酸为原料,温和条件下利用光催化合成该类化合物的方法仍然具有较高的实用价值.
本文以芳基酮酸及其衍生物作为自由基供体,通过蓝光照射,在光催化剂Ir(III)存在的条件下,得到苯甲酰基自由基,再发生加成环化反应,以良好的产率得到2-取代吲哚-3-乙酸酯类衍生物,反应如图 3 所示.

图3 由邻烯基芳基异腈合成吲哚-3-乙酸酯类衍生物
1 实验部分
1.1 仪器和试剂
1.1.1 主要试剂
2-碘苯胺,2-溴-6-甲基苯胺,2-溴-5-甲氧基苯胺,2-溴-4-三氟甲氧基基苯胺,2-溴-4-氟苯胺,2-溴-5-三氟甲基苯胺,4-氨基-3-溴苯甲酸甲酯,苯甲酰甲酸,对苯基苯乙酮,醋酸钯,丙烯酸乙酯,N,N-二甲基丙烯酰胺,三苯基膦,以上试剂均购买自上海毕得医药科技有限公司;甲酸,萨恩化学技术(上海)有限公司;乙酸酐,上海沃凯生物技术有限公司;DMF(N,N-Dimethylformamide,N,N-二甲基甲酰胺),广东光华科技股份有限公司;200~300目柱层析用硅胶,青岛海洋化工厂.
1.1.2 主要仪器
SGW X-4B 型显微熔点仪,上海仪电物理光学仪器有限公司;20W 蓝色LED实验灯,前方照明公司;AVANCE III 400 MHz型核磁共振仪,德国Bruker公司;Impact HD Q-TOF型高分辨质谱仪,德国Bruker公司;VECTOR II型光谱仪,德国Bruker公司;旋转蒸发仪RE-52AA,上海亚荣科技有限公司;加热磁力搅拌器MS-H-Pro+,SCILOGEX公司.
1.2 2-取代吲哚衍生物的合成
1.2.1 底物的合成
将邻溴苯胺衍生物(5.0 mmol)、丙烯酸乙酯(6.0 mmol)、醋酸钯(10 mol%)、三苯基磷(20 mol%)、三乙胺(30.0 mmol)、乙腈(10 mL)加入反应瓶中,90 ℃反应过夜.待反应完成后,冷却至室温,过滤并用乙酸乙酯洗涤.减压除去溶剂,快速柱层析(PE/EA=8∶1,体积比) 得到相对应的邻烯基苯胺衍生物.
将甲酸(6.0 mmol) 和乙酸酐(6.4 mmol)加入圆底烧瓶中,50 ℃反应2 h后冷却至室温得混合酸酐.邻烯基苯胺衍生物(4.0 mmol)溶于THF(Tetrahydrofuran,四氢呋喃,20 mL)中,冰浴下滴加混合酸酐.滴加完毕,将反应升至室温,继续搅拌2 h.反应完全后,加入饱和NaHCO3水溶液淬灭反应.乙酸乙酯萃取3次,合并有机相,无水硫酸钠干燥,过滤,并真空浓缩,重结晶得邻烯基甲酰苯胺衍生物.
将邻烯基甲酰苯胺衍生物(4.0 mmol)的THF(25 mL)溶液以及三乙胺(12 mmol)加入50 mL圆底烧瓶中,冰浴下缓慢滴加三氯氧磷(6.0 mmol).滴加完毕,保持冰浴继续反应2 h.反应完全后,加入饱和NaHCO3水溶液淬灭反应.乙酸乙酯萃取3次,合并有机相,无水硫酸钠干燥,过滤,减压除去溶剂,快速柱层析(PE/EA=8∶1,体积比)得到相应的2-乙烯基芳基异腈衍生物.
1.2.2 2-取代吲哚衍生物的合成
将2-乙烯基芳基异腈类衍生物(0.2 mmol)、苯甲酰甲酸(0.4 mmol)、[Ir(dF(CF3)ppy)2(dtb -bpy)]PF6(2 mol%),DMF(2 mL)依次加入10 mL透光瓶中,密封,20 W蓝光照射下搅拌12 h.反应完全后,加入饱和NaHCO3水溶液猝灭反应,乙酸乙酯萃取三次,无水硫酸钠干燥,减压除去溶剂后进行快速柱层析(PE/EA=8∶1,体积比)得到相对应吲哚衍生物,最后用核磁、红外、质谱、熔点仪对产物进行表征.
产品的表征数据如下:
2-(2-苯甲酰基吲哚-3-基)乙酸乙酯(3aa),黄色固体(31.3 mg,产率51%).熔点:138.4 ℃~139.5 ℃.1H NMR(400 MHz,CDCl3)δ8.98(s,1H,NH),7.83-7.77(m,2H,ArCH,ArCH),7.65(d,J=8.0 Hz,1H,ArCH),7.59(t,J=7.5 Hz,1H,ArCH),7.49(t,J=7.5 Hz,2H,ArCH,ArCH),7.41-7.33(m,2H,ArCH,ArCH),7.20-7.14(m,1H,ArCH),4.11(q,J=7.0 Hz,2H,EtCH2),3.82(s,2H,CH2),1.21(t,J=7.0 Hz,3H,EtCH3).13C NMR(101 MHz,CDCl3)δ189.03,171.10,139.24,136.55,132.46,132.29,128.96,128.84,128.54,126.67,121.31,121.11,116.52,112.31,61.11,31.50,14.39.IR:3 345,1 728,1 631,1 534,1 443.HRMS (ESI):calculated for C19H17NO3[M+H]+:308.128 1,found 308.129 0.
2-(2-苯甲酰基-7-甲基吲哚-3-基)乙酸乙酯(3ba),黄色固体(31.5 mg,产率49%).熔点:173.1 ℃~174.0 ℃.1H NMR(400 MHz,CDCl3)δ8.98(s,1H,NH),7.81-7.79(m,2H,ArCH,ArCH),7.63-7.57(m,1H,ArCH),7.49(t,J=7.5 Hz,3H,ArCH,ArCH,ArCH),7.16(d,J=7.0 Hz,1H,ArCH),7.12-7.07(m,1H,ArCH),4.09(q,J=7.0 Hz,2H,EtCH2),3.75(s,2H,CH2),2.51(s,3H,ArCH3),1.20(t,J=7.0 Hz,3H,EtCH3).13C NMR(101 MHz,CDCl3)δ189.21,171.03,139.38,136.43,132.42,132.22,128.94,128.81,128.22,126.90,121.65,121.40,118.93,116.89,61.08,31.71,16.78,14.38.IR:3 327,1 723,1 612,1 530,1 442.HRMS(ESI):calculated for C20H19NO3[M+H]+:322.143 8,found 322.1 450.
2-(2-苯甲酰基-6-甲氧基吲哚-3-基)乙酸乙酯(3ca),黄色固体(31.7 mg,产率47%).熔点:133.6 ℃~134.2 ℃.1H NMR(400 MHz,CDCl3)δ8.95(s,1H,NH),7.79-7.74(m,2H,ArCH,ArCH),7.58(t,J=7.5 Hz,1H,ArCH),7.52-7.46(m,3H,ArCH,ArCH,ArCH),6.85-6.78(m,2H,ArCH,ArCH),4.10(q,J=7.0 Hz,2H,EtCH2),3.85(s,3H,OMeCH3),3.75(s,2H,CH2),1.21(t,J=7.0 Hz,3H,EtCH3).13C NMR (101 MHz,CDCl3)δ188.35,171.08,160.08,139.57,137.91,132.12,131.64,128.79,128.77,123.14,122.34,117.36,112.94,93.74,55.73,31.62,14.39.IR:3 311,1 729,1 623,1 530,1 436.HRMS (ESI):calculated for C20H19NO4[M+H]+:338.138 7,found 338.139 6.
2-(2-苯甲酰基-5-三氟甲氧基吲哚-3-基)乙酸乙酯(3da),黄色固体(30.5 mg,产率39%).熔点:115.1 ℃~116.9 ℃.1H NMR(400 MHz,CDCl3)δ9.17 (s,1H,NH),7.82-7.75(m,2H,ArCH,ArCH),7.64-7.59(m,1H,ArCH),7.52-7.48(m,3H,ArCH,ArCH,ArCH),7.36(d,J=9.0 Hz,1H,ArCH),7.22-7.19(m,1H,ArCH),4.10(q,J=7.0 Hz,2H,EtCH2),3.79(s,2H,CH2),1.22(t,J=7.0 Hz,3H,EtCH3).13C NMR (101 MHz,CDCl3)δ188.61,170.66,143.58,138.56,134.45,133.65,132.58,128.86,128.70,128.34,120.57,120.72(q,J=257.0 Hz),116.15,113.24,113.14,61.10,31.18,14.09.IR:3 327,1 727,1 637,1 536,1 451.HRMS (ESI):calculated for C20H16F3NO4[M+H]+:392.110 4,found 392.111 5.
2-(2-苯甲酰基-5-氟吲哚-3-基)乙酸乙酯(3ea),黄色固体(30.0 mg,产率43%).熔点:170.5 ℃~170.9 ℃.1H NMR (400 MHz,DMSO)δ11.78(s,1H,NH),7.77-7.73(m,2H,ArCH,ArCH),7.71-7.67(m,1H,ArCH),7.58(t,J=7.5 Hz,2H,ArCH,ArCH),7.53-7.46(m,2H,ArCH,ArCH),7.21-7.16(m,1H,ArCH),4.00(q,J=7.0 Hz,2H,EtCH2),3.88(s,2H,CH2),1.12(t,J=7.0 Hz,3H,EtCH3).13C NMR(101 MHz,DMSO)δ188.23,170.53,157.23 (d,J=235.0 Hz),138.42,133.42,133.20,132.44,128.80,128.68,127.78(d,J=10.0 Hz),115.32(d,J=5.5 Hz),114.33(d,J=27.0 Hz),114.20(d,J=10.0Hz),104.91(d,J= 23.0Hz),60.14,40.14,39.93,39.72,39.51,39.30,39.09,38.89,30.38,14.02.IR:3 312,1 724,1 610,1 531,1 447.HRMS (ESI):calculated for C19H16FNO3[M+H]+:326.118 7,found 326.119 8.
2-(2-苯甲酰基-6-三氟甲基吲哚-3-基)乙酸乙酯(3fa),黄色固体 (36.0 mg,产率48%).熔点:152.1 ℃~153.2 ℃.1H NMR(400 MHz,CDCl3)δ9.36(d,J=22.0 Hz,1H,NH),7.81-7.79(m,2H,ArCH,ArCH),7.73(d,J=8.5 Hz,1H,ArCH),7.68-7.60(m,2H,ArCH,ArCH),7.51(t,J=7.5 Hz,2H,ArCH,ArCH),7.37(d,J=8.5 Hz,1H,ArCH),4.11(q,J=7.0 Hz,2H,EtCH2),3.83(s,2H,CH2),1.22(t,J=7.0 Hz,3H,EtCH3).13C NMR (101 MHz,CDCl3)δ188.94,170.98,138.62,135.20,134.33,132.97,130.43,129.13,128.96,128.20(q,J=32.3 Hz),124.80(q,J=273.0 Hz,CF3),121.90,117.50(q,J=4.0 Hz),115.86,110.15(q,J=5.0 Hz),61.33,31.32,14.36.IR:3 317,1 714,1 630,1 538,1 440.HRMS (ESI):calculated for C20H16F3NO3[M+H]+:376.115 5,found 376.116 3.
2-苯甲酰基-3-(2-乙氧基-2-氧代乙基)-5-吲哚甲酸甲酯(3ga),黄色固体(32.9 mg,产率45%).熔点:166.5 ℃~167.9 ℃.1H NMR(400 MHz,CDCl3)δ9.38(s,1H,NH),8.43(s,1H,ArCH),8.02-7.99(m,1H,ArCH,),7.81-7.76(m,2H,ArCH,ArCH,),7.61(t,J=7.5 Hz,1H,ArCH),7.50(t,J=7.5 Hz,2H,ArCH,ArCH),7.39(d,J=9.0 Hz,1H,ArCH),4.12(q,J=7.0 Hz,2H,EtCH2),3.93(s,3H,CO2MeCH3),3.83(s,2H,CH2),1.23(t,J=7.0 Hz,3H,EtCH3).13C NMR (101 MHz,CDCl3)δ188.85,170.88,167.71,138.81,133.57,132.75,128.99,128.90,128.12,127.34,124.54,123.18,117.58,112.16,61.30,52.23,31.31,14.34,(one carbon missing due to overlap).IR:3 317,1 714,1 630,1 538,1 440.HRMS(ESI):calculated for C21H19NO5[M+H]+:366.113 6,found 366.134 5.
2-(2-苯甲酰基吲哚-3-基)-N,N-二甲基乙酰胺(3ha),黄色固体(22.6 mg,产率37%).熔点:159.4 ℃~160.5 ℃.1H NMR (400 MHz,CDCl3)δ9.01 (s,1H,NH),7.77-7.71(m,3H,ArCH,ArCH,ArCH),7.58(t,J=7.5 Hz,1H,ArCH),7.47(t,J=7.5 Hz,2H,ArCH,ArCH),7.35-7.28(m,2H,ArCH,ArCH),7.15-7.11(m,1H,ArCH),3.86(s,2H,CH2),2.90(s,3H,NMeCH3),2.83(s,3H,NMeCH3).13C NMR (101 MHz,CDCl3)δ189.01,170.44,139.56,136.79,132.22,131.76,128.99,128.90,128.70,126.62,122.02,121.02,118.46,112.23,37.44,35.88,31.32.IR:3 256,1 634,1 530,1 444.HRMS (ESI):calculated for C19H18N2O2[M+H]+:307.144 1,found 307.144 5.
2-(2-对苯基苯甲酰基吲哚-3-基)乙酸乙酯(3ab),黄色固体(25.3 mg,产率33%).熔点:93.4 ℃~95.3 ℃.1H NMR(400 MHz,CDCl3)δ9.00 (s,1H,NH),7.92-7.87(m,2H,ArCH,ArCH),7.72(d,J=8.0 Hz,2H,ArCH,ArCH),7.69-7.63(m,3H,ArCH,ArCH,ArCH),7.51-7.46 (m,2H,ArCH,ArCH),7.44-7.39(m,2H,ArCH,ArCH),7.38-7.34(m,1H,ArCH),7.20-7.16(m,1H,ArCH),4.12(q,J=7.0 Hz,2H,EtCH2),3.90(s,2H,CH2),1.20(t,J=7.0 Hz,3H,EtCH3).13C NMR (101 MHz,CDCl3)δ188.39,170.98,145.16,139.88,137.66,132.21,129.55,129.01,128.35,128.25,127.27,126.43,121.11,120.93,116.20,112.13,60.94,31.40,14.21.IR:3 345,1 729,1 630,1 534,1 441.HRMS (ESI):calculated for C25H21NO3[M+H]+:384.159 4,found 384.160 1.
2 结果与讨论
2.1 条件优化
在大量文献调研的基础上,本文以2-异腈基肉桂酸乙酯和苯甲酰甲酸为模板底物,通过对催化剂和溶剂的筛选对反应进行优化.
首先对[Ir(dtb-bpy)(ppy)2]PF6、[Ir(dF(CF3)ppy)2(dtb-bpy)]PF6、[Ir(dF(CF3)ppy)2(1,10-Phen)]PF6、Ir(ppy)3(催化剂结构如图4所示),以及Eosin Y(曙红Y) 等光催化剂进行了考察.实验结果表明,Ir类光催化剂均能促进该反应的进行,以17%~51% 的收率得到目标产物3aa(如表1所示).

图4 吲哚-3-乙酸酯的合成及光催化剂结构示意图

表1 反应条件优化
注:①a:加入K2CO3(2 equiv);b:加入K2HPO4(2 equiv);c:5 W Blue LED. ②反应条件:1a(0.2 mmol),2a(0.4 mmol),PC(2 mol %),DMF(2 mL),20W Blue LED,12 h.
通过单晶衍射对产物3aa结构进行确认,晶体结构图如图5所示,精修参数如表2所示.处理方法为,在室温条件下,将化合物3aa的晶体置于显微镜下,选取形状规则光亮尺寸合适的单晶,置于X-射线单晶衍射仪上,经过石墨单色器单色纯化的Mo-Kα射线(λ=0.710 73 nm) 照射,通过ω扫描方式收集上述单晶的衍射数据,所收集的数据运用SAINT程序进行数据还原,然后用SADABS程序做吸收校正.单晶结构使用SHELXS-97程序采用直接法进行初始结构解析,使用SHELXL-97程序进行全矩阵最小二乘法精修,对骨架中所有的非氢原子进行各项异性精修.骨架结构中的氢原子位置全部采用理论加氢的方式进行确定,所有的氢原子则采用各向同性精修.
在三种Ir类催化剂中,[Ir(dF(CF3)ppy)2(dtb-bpy)]PF6表现最好,因此本文在该催化剂的参与下,考察了常见有机溶剂对反应结果的影响.结果显示,当DMF作溶剂时,反应较好,以51%的收率得到目标产物,而其它溶剂则给出较差的反应结果.根据文献[16]报道,碱能够促使脱羧反应进行得到自由基,因此,为进一步提高产物收率,本文考察了碱对反应的影响.令人失望的是,K2CO3或者K2HPO4的加入并没有对产率有所帮助,这个结果说明苯甲酰甲酸可直接经由单电子转移,得到阳离子自由基,进而完成脱羧产生自由基.改变蓝光灯功率对反应未造成影响,故选用易购买且易操作的20 W LED蓝光灯泡完成反应.最终,本文确定了最优条件为:1a(0.2 mmol),2a(0.4 mmol),[Ir(dF(CF3)ppy)2(dtb-bpy)]PF6(2 mol%),以DMF(2 mL)为溶剂在20 W蓝光照射下反应12 h.
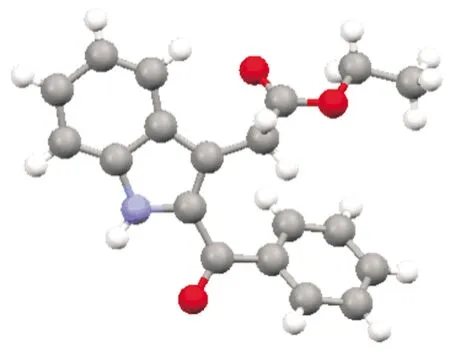
图5 化合物3aa的晶体结构图
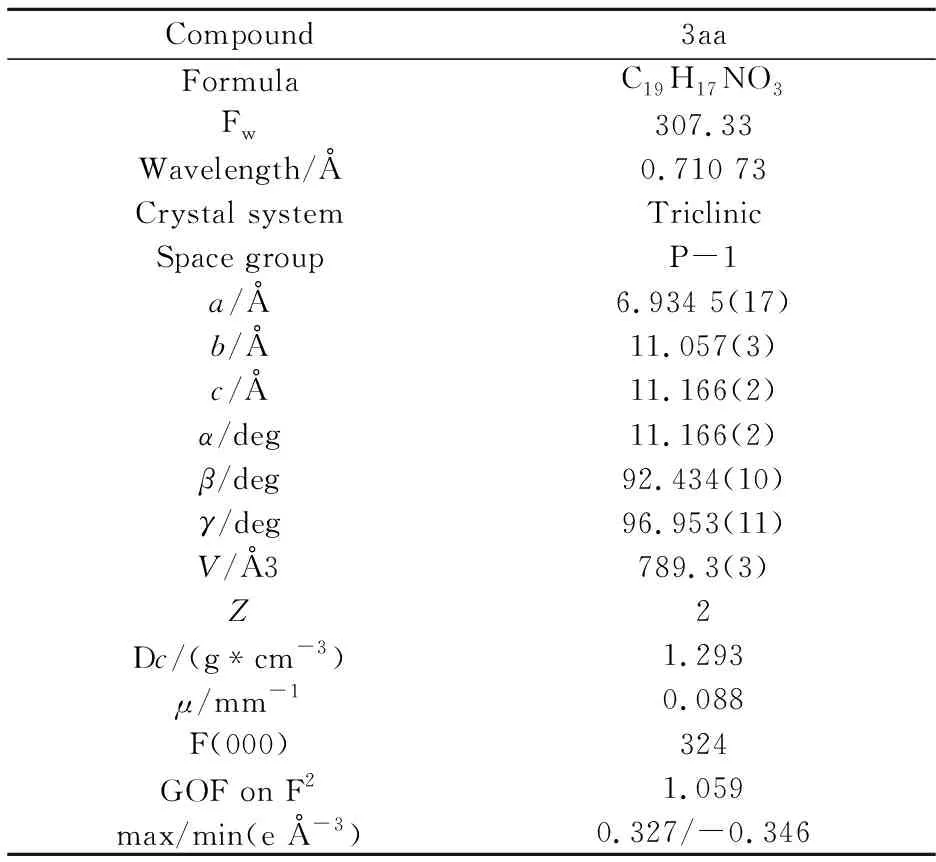
表2 化合物3aa的晶体结构与精修参数
2.2 底物适用性考察
在最优条件下,本文对反应的底物适用性进行了考察.如图6所示,空间位阻对该反应几乎没有影响.比如在异腈邻位引入甲基时,仍然可以以49%的收率得到目标产物.
然后,本文对电子效应进行了考察.反应结果表明,该反应对吸电子和给电子基团都表现出了很好的耐受性.在芳环上引入氟,甲氧基,三氟甲基和三氟甲氧基时,都可以以中等以上的收率得到目标产物.
再进一步,本文考察了功能化基团对反应的影响,发现将酯基引入反应体系中,仍能够以45%的收率得到目标产物,这为反应的衍生化提供的可能性.当将3-位酯基替换为酰胺片段时,反应产量明显下降,只能以37%的收率得到产物,这可能是因为酰胺的氮原子对自由基有一定的猝灭作用.
最后,本文将苯基酮酸替换成联苯基酮酸时,产率下降为33%,这可能是由于芳基酮酸中苯基的引入增加了酰基自由基生成的难度.
2.3 控制实验与可能性反应机理
为了研究反应机理,在标准反应条件下,分别进行了避光和不加光催化剂的实验,如图7所示,均未观测到产物的生成,说明光照和光催化剂对于该反应的进行都是必不可少的条件.另外,在标准反应条件下,当自由基抑制剂TEMPO (2,2,6,6 -tetramethylpiperidine-1-oxyl,2,2,6,6- 四甲基哌啶氧化物) 加入至0.1 mmol 时,反应被部分抑制;加入至0.2 mmol时,反应被完全抑制无法进行,如表3所示,说明该反应可能经历自由基过程.
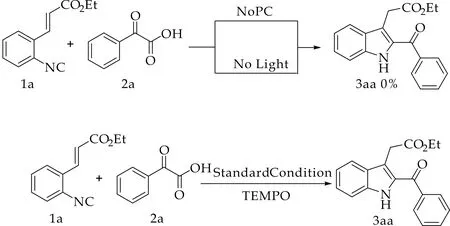
图7 机理探究实验

表3 自由基抑制实验研究
结合控制实验结果与相关文献的报道[17,18],本文推测该反应可能按照以下途径(图8) 进行:首先,光催化剂在光照下生成激发态的*Ir3+,进一步α-酮酸的单电子氧化,使其发生自由基脱羧得到苯甲酰基自由基与Ir2+.苯甲酰基自由基与1a中异腈的碳氮三键发生自由基加成得到中间体A,继而与邻位的烯基反应得到中间体B.烷基自由基B可以将Ir2+氧化为Ir3+完成光催化循环,其自身被还原为碳负离子中间体C.最后中间体C得到质子并异构化得到最终产物3aa.

图8 可能的反应机理
3 结论
本文开发了一种利用光催化自由基环化合成吲哚-3-乙酸酯类衍生物的新方法.利用该策略,本文以良好的产率得到九种2-取代吲哚衍生物.该方法条件温和,对环境友好且具有良好的底物适用性,为吲哚-3-乙酸酯类化合物的合成提供新的途径,同时也为光诱导的自由基加成环化反应提供了新的思路.
猜你喜欢
可再生能源(2022年5期)2022-06-09
中国典型病例大全(2022年10期)2022-05-10
城市道桥与防洪(2022年3期)2022-05-08
湖北农机化(2021年11期)2021-07-01
安全与环境工程(2021年2期)2021-04-02
中国应急管理科学(2021年9期)2021-03-16
人物画报(2020年29期)2020-03-14
人物画报(2020年36期)2020-03-13
中学生数理化·高二版(2016年3期)2016-12-26
安徽医科大学学报(2015年9期)2015-12-16
