
黔产民族药骨碎补药材HPLC指纹图谱研究*
2020-05-08 12:00黄春江徐文芬黄侨宗吴付玉肖仕林林雨鑫
贵州科学 2020年1期
高 月,黄春江,徐文芬,黄侨宗,吴付玉,肖仕林,林雨鑫
(贵州中医药大学药学院,贵州 贵阳 550025)
骨碎补为水龙骨科植物槲蕨Drynariafortunei(Kunze)J.Sm.的干燥根茎,具有疗伤止痛、补肾强骨的功效,用于治疗跌扑闪挫、筋骨折伤、肾虚腰痛和筋骨痿软等[1]。现代研究表明骨碎补中化学成分主要有黄酮类、酚酸类、木脂素、甾体类及三萜类等;具有抗骨质疏松、抗炎、促进骨折愈合、促进牙齿生长、降血脂等作用[2]。目前,骨碎补药材的质量控制和质量评价方法,除2015年版《中国药典》一部收载骨碎补质量标准中,以柚皮苷作为药材有效性控制的指标成分外,文献报道还有以柚皮苷、新北美圣草苷及其他指标成分作为其质量评价指标[3-5]。然而,中药的疗效并非某一个或几个成分作用的结果,建立符合中药特点的质量控制和评价方法是亟待解决的问题。中药指纹图谱能较全面地反映药材所含化学成分的相对关系,能真正对中药内在质量进行有效表征、综合评价和全面控制[6-7]。本文采用高效液相色谱法,对12个不同批次的黔产骨碎补药材进行指纹图谱测定分析,从整体观念出发,以多组分角度更客观、更有效地反映该药材的内在质量,为骨碎补药材的质量综合评价以及相关制剂的质量控制提供参考。
1 仪器与材料
1.1 仪器
UltiMate 3000型高效液相色谱仪(美国Thermo Fisher公司)、AG135型十万分之一电子分析天平(瑞士Mettler-Toledo公司)、SB25-12DTD型超声提取器(宁波新艺超声设备有限公司)等。
1.2 材料
柚皮苷对照品(批号:110722-201714,中国食品药品检定研究院)、新北美圣草苷(批号:BCY-001524,江西佰草源生物科技有限公司);乙腈为色谱纯(美国Tedia公司),无水乙醇、甲醇、醋酸为分析纯(均为天津市富宇精细化工有限公司),试验用水为重蒸馏水。供试骨碎补药材经贵州中医药大学孙庆文教授鉴定为水龙骨科槲蕨DrynariaroosiiNakaike的干燥根茎,12批黔产样品产地信息详见表1。
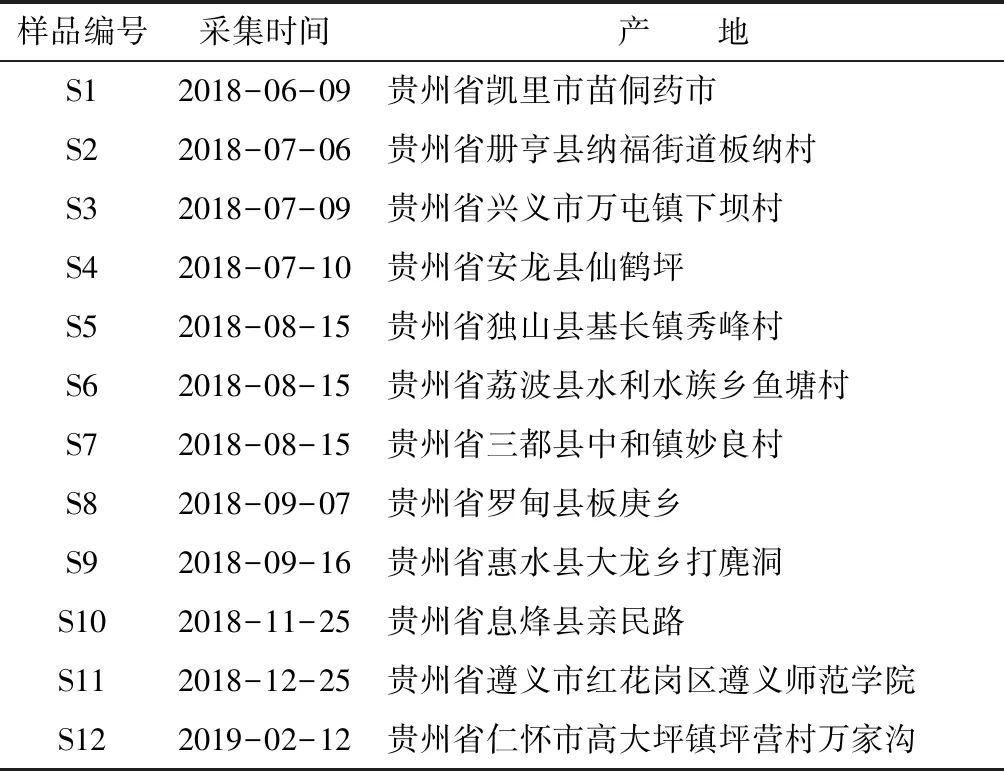
表1 12批骨碎补样本采集信息
2 方法与结果
2.1 对照品溶液制备
精密称取新北美圣草苷0.5 mg、柚皮苷0.3 mg,分别置于10 mL量瓶中,加甲醇溶解并定容至刻度线,摇匀,即得对照品溶液。
2.2 供试品溶液制备
取骨碎补药材粉末(过三号筛)约0.5 g,精密称定,置锥形瓶中,精密加入70%乙醇20 mL,称定重量,超声处理(功率500 W,频率40 kHz)30 min,放冷至室温,用70%乙醇补足减失的重量,取续滤液,即得供试品溶液。
2.3 色谱条件
色谱柱为Diamonsil-C18(250 mm×4.6 mm,5 μm),流动相为乙腈(A)-0.5%的醋酸水溶液(B),采用梯度洗脱:0~10 min,2%~8%A;10~15 min,8%~15%A;15~22 min,15%~20%A;22~35 min,20%~22%A;35~45 min,22%~90%A;流速1.0 mL/min,检测波长为260 nm,柱温为30 ℃,进样量为10 μL。
2.4 方法学考察
2.4.1 图谱完整性考察
取供试品溶液制备的提取溶剂70%乙醇溶液,按2.3色谱条件下进样检测,结果显示提取溶剂及流动相相对指纹图谱测定没有干扰。另取S9样品(贵州惠水),按2.2项下方法制备供试品溶液,按2.3色谱条件下进样检测,考察指纹图谱记录的完整性,记录45 min后以梯度结束时的流动相比例继续洗脱,并记录色谱图至90 min,结果显示50 min以后无色谱峰出现,故确定采集图谱时间为45 min,结果见图1。
2.4.2 精密度试验
取S9(贵州惠水)样品,按2.2项下方法制备供试品溶液1份,按2.3色谱条件下进样检测,连续进样6次,记录色谱图,以柚皮苷为参比峰,计算各共有峰相对保留时间的RSD均小于0.15%,各相对峰面积的RSD均小于1.5%,表明仪器的精密度良好。
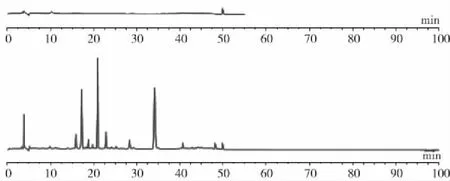
图1 指纹图谱完整性考察结果Fig.1 Investigation results of fingerprint integrity
2.4.3 重复性试验
取S9(贵州惠水)样品6份,按2.2项下方法制备供试品溶液,按2.3色谱条件下进样检测,记录色谱图,以柚皮苷为参比峰,计算各共有峰相对保留时间的RSD均小于0.42%,相对峰面积的RSD均小于2.6%,结果表明方法重复性良好。
2.4.4 稳定性试验
取S9(贵州惠水)样品1份,按2.2项下方法制备供试品溶液,按2.3色谱条件下分别在0 h、2 h、4 h、8 h、16 h、24 h进样检测,记录色谱图,以柚皮苷为参比峰,计算各共有峰相对保留时间的RSD均小于0.28%,结果,相对峰面积的RSD均小于2.6%,表明供试品溶液在24 h内稳定性良好。
2.5 样品测定
取12批骨碎补样品,按2.2项下方法制备供试品溶液,按2.3项下色谱条件进样测定,记录测得的原始图谱。
2.6 指纹图谱的建立
将所得色谱图分别导入中药色谱指纹图谱相似度评价系统(2004 A版)进行处理,以S9作为参照指纹图谱,选择中位数法作为对照指纹图谱的生成方法,时间宽度设为0.50,对色谱峰进行全谱峰匹配,并生成对照指纹图谱R,得到12批骨碎补药材的HPLC指纹图谱原始图谱叠加图及对照指纹图谱R(图2)。从12批药材指纹图谱共标定10个共有峰,其中8号共有峰、9号共有峰的保留时间分别与新北美圣草苷对照品和柚皮苷对照品的保留时间一致,且其紫外吸收光谱也一致,因此,指认8号共有峰为新北美圣草苷,9号共有峰为柚皮苷,并设定9号共有峰为参照峰,计算不同批次骨碎补指纹图谱中各共有峰的相对保留时间RSD在0.02%~0.23%之间,各共有峰的相对峰面积RSD在25.20%~126.31%之间,说明不同批次的骨碎补药材化学成分相对含量存在较大差异,结果见图3和表2、表3。
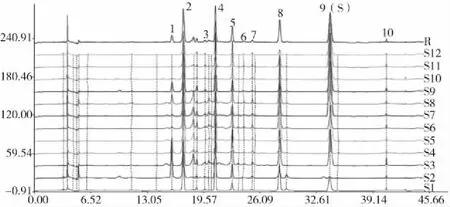
图2 12批骨碎补药材HPLC指纹图谱原始图谱叠加图及对照指纹图谱RFig.2 HPLC fingerprint,original fingerprint,superposition map,and control fingerprint R of the 12 batches of Drynariae Rhizoma
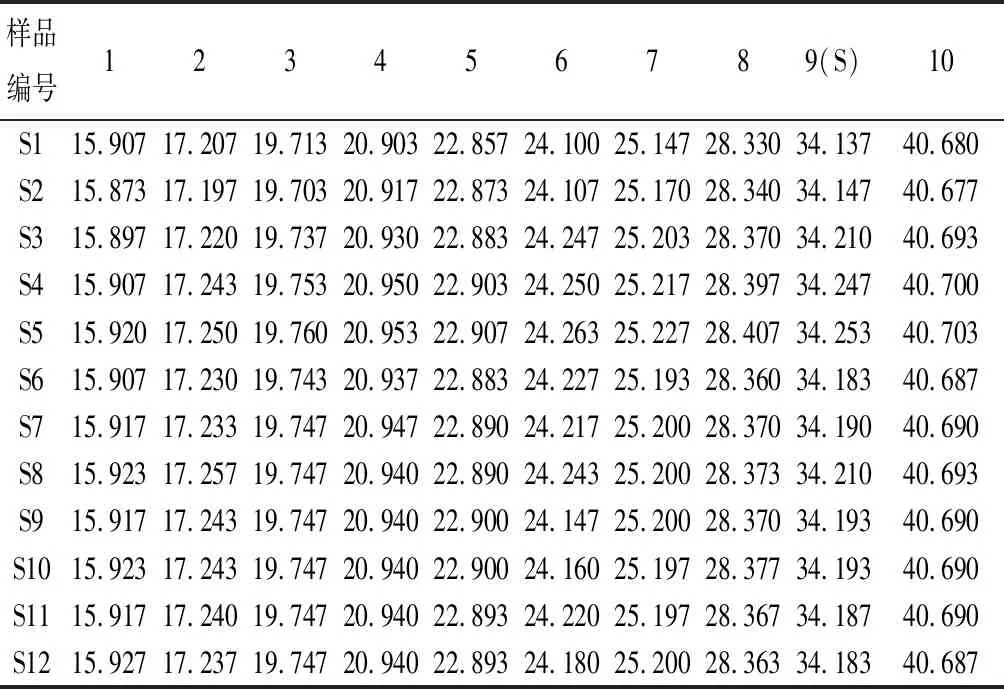
表2 12批骨碎补药材10个共有峰的相对保留时间
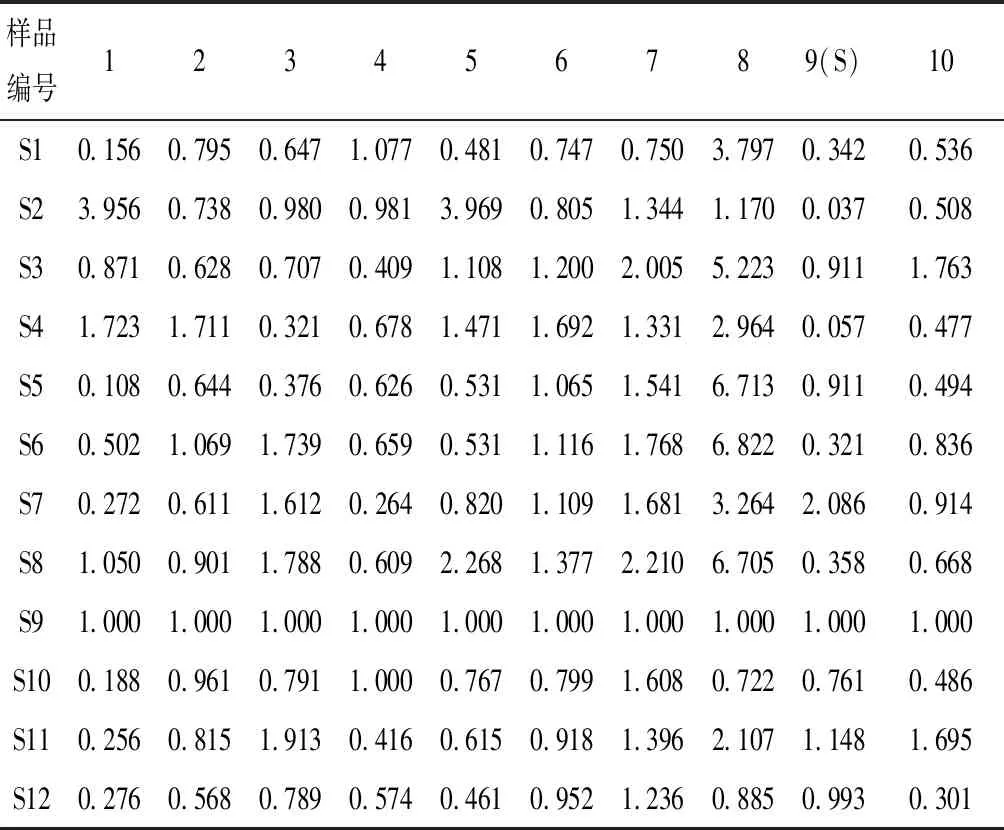
表3 12批骨碎补药材10个共有峰的相对峰面积
2.7 结果分析
2.7.1 相似度评价结果
采用指纹图谱软件对12批骨碎补药材进行相似度评价的结果为:各批次样品指纹图谱的相似度范围为0.551~0.957。由此可见,各批次骨碎补药材的化学成分及其含量有一定差别,结果见表4。
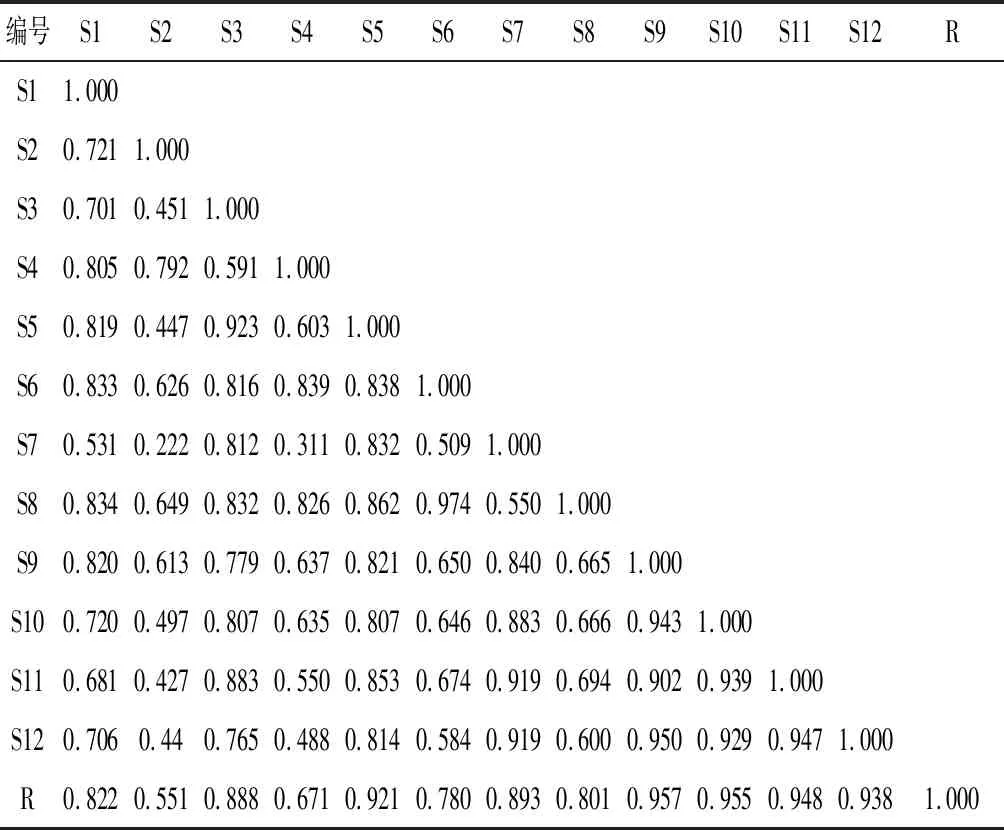
表4 12批骨碎补药材指纹图谱相似度计算结果
2.7.2 聚类分析结果
将12批不同批次骨碎补药材指纹图谱共有峰峰面积导入SPSS20.0软件进行聚类分析。运用组间连接法进行系统聚类分析,其中样品间距离的计算采用欧式平方距离法,结果见图4。结果显示:当判别条件距离为20时,大致分为三大类,S4(贵州省安龙县)可单独聚为一类;S3(贵州省兴义市)、S6~S8和S11(贵州省遵义市)聚为一类;S1~S2、S5(贵州省独山县)、S9~S10、S12(贵州省仁怀市)聚为一类。
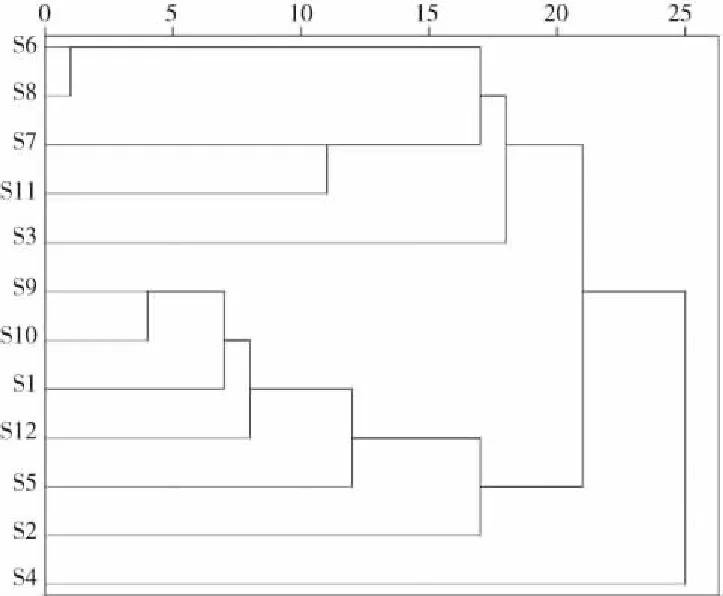
图4 12批骨碎补药材指纹图谱聚类树状关系图Fig.4 Cluster tree diagram of fingerprints of the 12 batches of Drynariae Rhizoma
2.7.3 主成分分析结果
采用SPSS20.0软件,将12批骨碎补药材指纹图谱的10个共有峰峰面积原始数据进行标准化处理,以主成分的特征值及贡献率为依据,进行主成分分析,计算相关系数矩阵、主成分特征值、累积贡献率并计算主成分综合得分[8],对12批骨碎补药材进行评价。根据因子分析结果可得总方差解释表,对各变量进行因子提取和因子旋转的结果,见表5和图5。
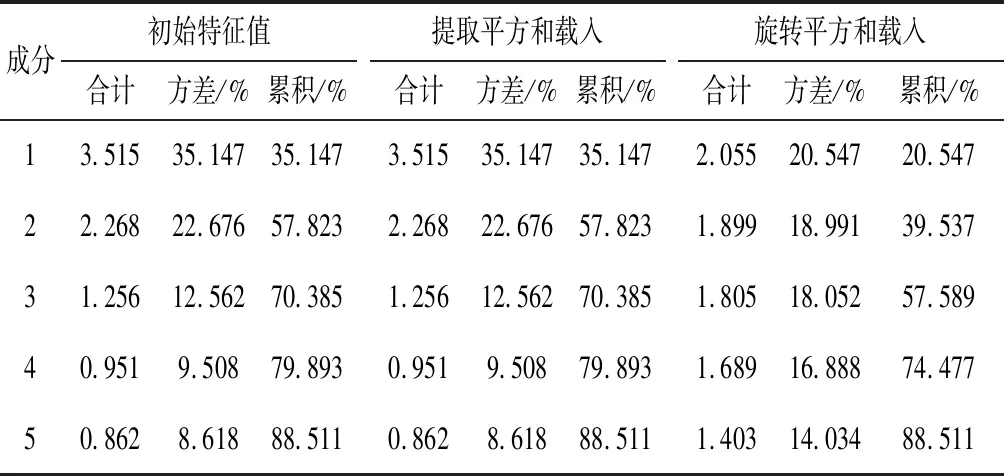
表5 骨碎补总方差解释变异量
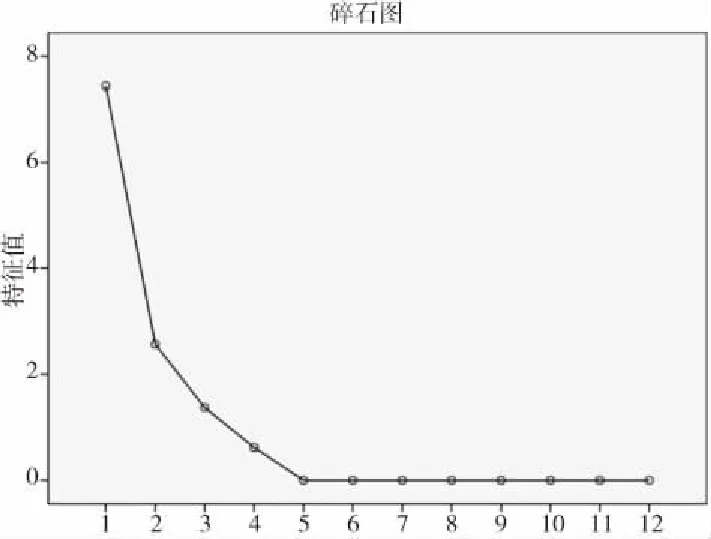
图5 主成分分析碎石图Fig.5 Principal component analysis gravel map
根据各因子特征值大于1的原则及因子的方差累计贡献率达到85%以上作为主成分提取的标准并结合表4,本试验需要提取的主成分主要有5个,其中提取主成分1的特征值为3.515,其贡献率为35.147%;提取主成分2的特征值为2.268,贡献率为22.676%;提取主成分3的特征值为1.256,贡献率为12.562%;提取主成分4的特征值为0.951,贡献率为9.508%;提取主成分5的特征值为0.862,贡献率为8.618%。这5个主成分的累积方差总贡献率达到88.511%,因此提取前5个主成分进行分析较为合适。进行正交旋转,得到不同批次骨碎补10个指标在5个主成分中的旋转成分矩阵,见表6。
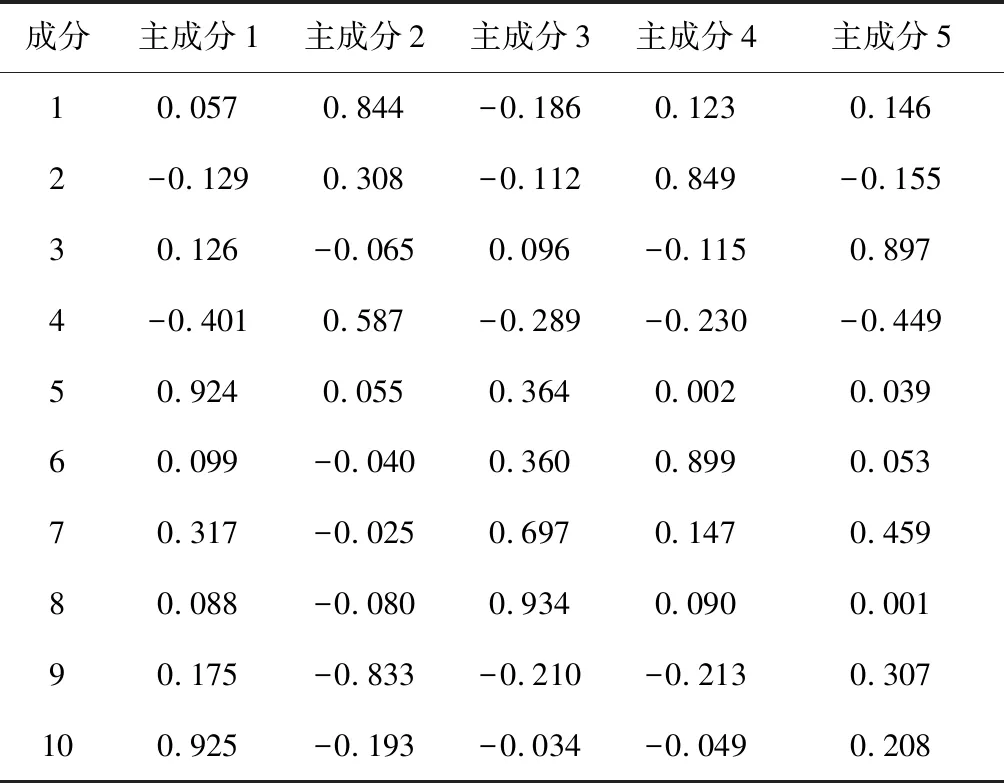
表6 黔产骨碎补旋转成分矩阵
根据表5、表6,以Y1,Y2,Y3,Y4,Y5代表5个主成分来作为12批次骨碎补成分所表达的信息进行评价,得出如下成分的线性关系表达式分别为Y1=0.057F1-0.129F2+0.126F3-0.401F4+0.924F5+0.099F6+0.317F7+0.088F8+0.175F9+0.925F10,Y2=0.844F1+0.308F2-0.065F3+0.587F4+0.055F5-0.040F6-0.025F7-0.080F8-0.833F9-0.193F10,Y3=-0.186F1-0.112F2+0.096F3-0.289F4+0.364F5+0.360F6+0.697F7+0.934F8-0.210F9-0.034F10,Y4=0.123F1+0.849F2-0.115F3-0.230F4+0.002F5+0.899F6+0.147F7+0.090F8-0.213F9-0.049F10,Y5=0.146F1-0.155F2+0.897F3-0.449F4+0.039F5+0.053F6+0.459F7+0.001F8+0.307F9+0.208F10[9]。表达式中各变量不是原始变量,而是标准化变量。
将上述所得表达式,与所对应的5个主成分的方差贡献率做内积,得到黔产骨碎补质量综合函数表达式:
Y综合得分=(Y1×35.147+Y2×22.676+Y3×12.562+Y4×9.508+Y5×8.618)/88.511
根据以上表达式得到12批次骨碎补综合得分值及排序,结果见表7。
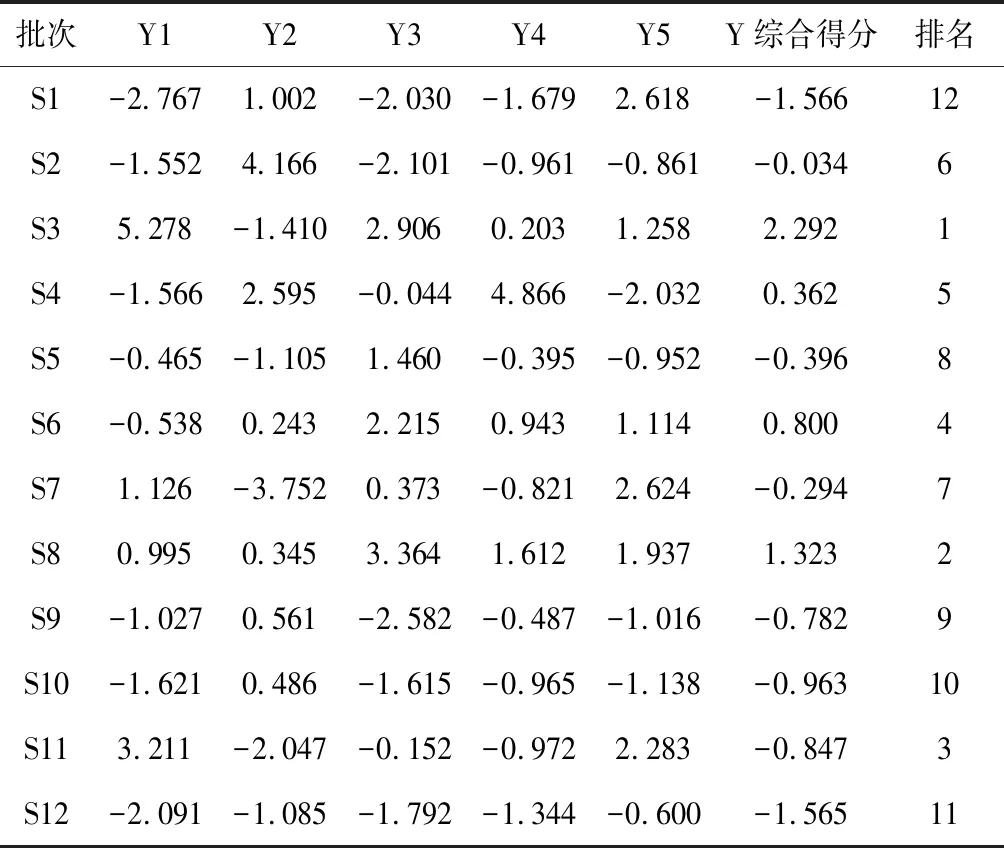
表7 12批次骨碎补主成分得分及综合得分排名
综合得分越高,表明质量越好[10-11]。12批黔产骨碎补综合得分排名由高到低依次为S3,S8,S11,S6,S4,S2,S7,S5,S9,S10,S12,S1。当选取第1、2、3个因子主成分分析,S1、S2、S9、S10相聚在一类,S3、S5、S6、S7、S8、S11聚为一类,S4、S12偏离较远,主成分分析与聚类分析、相似度评价结果基本一致,但其中S5在主成分分析与聚类分析两种模式识别结果中的差异较大。结果见图6。
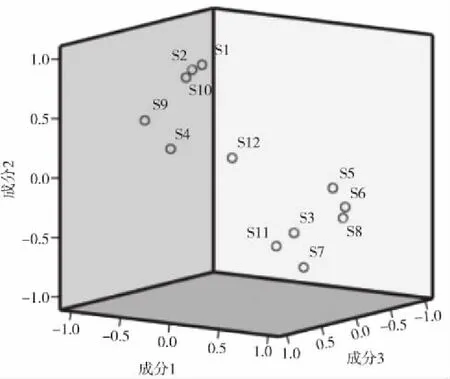
图6 选取第1、2、3个因子主成分分析结果Fig.6 Principal component analysis results of the first,second and third factor
3 讨论
本文以指纹图谱中主要色谱峰峰面积及色谱峰峰数为指标,考察了不同提取方法(超声、回流)、提取时间(15 min、30 min、45 min、60 min及75 min)、不同提取溶剂(70%甲醇、50%乙醇、60%乙醇、70%乙醇、80%乙醇及90%乙醇)对结果的影响。最终确定骨碎补指纹图谱测定的提取方法为40倍体积的70%乙醇超声提取30 min一次。
对不同流动相(甲醇-0.1%醋酸水、乙腈-0.2%磷酸水、乙腈-0.2%醋酸水、乙腈-0.5%醋酸水及乙腈-1%醋酸水)梯度洗脱考察结果表明,乙腈-0.5%醋酸水作为流动相峰形尖锐,分离度好且基线平稳;不同色谱柱Accucore C18(150 mm×4.6 mm,2.6 μm)、Hypersil GOLD(250 mm×4.6 mm,5 μm)和Diamonsil-C18(250 mm×4.6 mm,5 μm)考察结果表明,Diamonsil-C18(250 mm×4.6 mm,5 μm)色谱柱分离效果好;不同检测波长(254 nm、260 nm、283 nm)比较结果表明,260 nm处色谱峰多且信息完全。因此,确定色谱条件以Diamonsil-C18(250 mm×4.6 mm,5 μm)为色谱柱,检测波长为260 nm,流动相为乙腈-0.5%醋酸水梯度洗脱。
通过对12批黔产骨碎补药材样品的指纹图谱分析可知,其共有峰的相对保留时间差异较小,表明本文所建立的骨碎补药材指纹图谱测定方法重现性良好,但不同批次黔产骨碎补药材相对峰面积差异较大,表明该药材质量受地域影响较大。研究结果为完善黔产骨碎补药材的综合质量控制评价体系提供了数据参考。
猜你喜欢
煤化工(2022年3期)2022-07-08
数学物理学报(2021年6期)2021-12-21
数学物理学报(2021年5期)2021-11-19
数学物理学报(2021年3期)2021-07-19
色谱(2021年7期)2021-06-07
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
军事运筹与系统工程(2020年2期)2020-11-16
军事运筹与系统工程(2018年3期)2018-03-26
山东工业技术(2016年10期)2016-09-06
中亚信息(2016年10期)2016-02-13
