
TLR4在青光眼视神经损伤机制中的作用
2020-05-08 10:58张立宇许育新
国际眼科杂志 2020年5期
李 倩,张立宇,许育新
青光眼是一种以视乳头萎缩凹陷、视野缺损及视力下降为共同特征的不可逆的致盲性疾病,其视神经损伤的本质为视神经节细胞的凋亡。尽管通过药物干预和手术控制眼压可以对青光眼起到一定的治疗作用,但如何从根本上阻止青光眼的进一步发展仍处于探索阶段。因此,研究青光眼视神经损伤机制,通过阻断视神经损伤而治疗青光眼至关重要。近几年,免疫机制对青光眼视神经损伤的影响成为研究热点,本文中主要对Toll样受体4(TLR4)通过不同免疫通路并与神经胶质细胞相互作用引起青光眼患者视神经损伤进行综述。
0 引言
青光眼是一组以视网膜神经节细胞(retinal ganglion cells, RGCs)的逐渐凋亡和轴突变性,以及视野的缺损为特点的神经退行性疾病,病理性眼压升高是其主要危险因素。青光眼是世界性导致人类不可逆失明的主导因素, 其发病率高达2.6%[1-2]。迄今为止,全球40~80岁的青光眼患者约占总人口的3.5%,而青光眼患者的人数至2040年预计将增加到111万人[3]。现阶段青光眼的治疗以传统的降眼压和视神经保护药物以及滤过性手术和激光手术为主要方法[4]。然而,正常眼压的青光眼患者或者是眼压控制良好的青光眼患者视神经损伤仍然存在。由此可见,青光眼视神经损伤的机制尚未完全明了。研究青光眼患者视神经损伤的具体机制,通过阻断或者延缓受损机制从根本上解决视神经损伤的发生尤为重要。免疫应答与神经退行性疾病(例如阿尔兹海默症、帕金森疾病)息息相关,而Toll样受体4(TLR4)作为免疫应答中信号转导的重要部分,其对神经退行性疾病的影响逐渐受到大家的关注[5]。目前,已有多篇报道发现TLR4和神经胶质细胞的激活在阿尔兹海默症中的病理作用[5-6],而青光眼作为神经退行性疾病的一种,其视神经损伤机制与TLR4亦密不可分。本文将就TLR4通过免疫作用导致青光眼视神经损伤进行综述。尽管青光眼在临床上分为不同类型,但都以高眼压为其主要的高危因素,导致视神经损伤的机制也大致相同,本文中讨论的青光眼视神经损伤指的是所有病理性高眼压导致的视神经损伤。
1 青光眼视神经损伤的机制
关于青光眼的视神经损伤机制主要有两种学说,即机械学说和缺血学说。机械学说强调视神经受压,轴浆流中断的重要性。缺血学说则强调视神经供血不足,对眼压耐受性的重要性。目前一般认为青光眼视神经损害的机制是机械压迫和缺血的共同作用。在高眼压作用下,视神经纤维和巩膜相互施压以致变形,视神经轴突萎缩,轴浆流受阻,影响视网膜神经节细胞(retinal ganglion cells,RGCs)的营养供应,使视神经纤维坏死、溶解、消失[7]。正常眼部血管具有自我调节能力,使血流相对稳定,当视盘灌注压明显下降,眼压升高导致微循环自身调节功能紊乱,通过形成缺血性环境加剧视神经损伤。随着对各种细微分子的研究逐渐加深,各种机制也随之被提出。Qu 等[8]通过分析青光眼患者视神经节细胞的凋亡过程,将机制分为通过受体依赖的胱天蛋白酶(caspase)内部途径和外部途径以及激发该家族相关分子促进凋亡三个方面。通过该三个方面详细解释了神经营养因子缺乏、神经胶质细胞激活、氧化应激、自噬作用、细胞因子、谷氨酸毒性作用和钙离子作用等机制的具体过程。此外,国内有学者通过宏观因素和微观因素来区分作用机制,或者是强调轴突相关机制和神经胶质细胞免疫应答在青光眼中的重要影响[9-10]。Evangelho等[11]从缺氧、缺血、兴奋性毒性、视轴传导和营养障碍多方面总结了视神经损伤的机制。尽管视神经损伤的机制有多种,但均表现为视神经节细胞的死亡。近年来越来越重视青光眼发病与免疫调节存在的相关性[12]。多种机制通过免疫调节导致神经节细胞死亡。例如氧化应激产生的氧化物或者氮化物可通过激活P38MAPK,来激活caspase-3而引起神经节细胞凋亡损伤。而其详细的免疫炎症通路皆与TLR4有着密不可分关系。

图1 TLR4通过 MyDD8 依赖途径损伤视神经。
2 Toll样受体
Toll样受体(toll like receptor,TLR)是一类典型的1型模式识别受体(pattern recognition receptors,PRR),在天然免疫系统中发挥着重要的作用[13-14],该家族共有13个成员(TLR1-13)被发现,其中10个与人有关[15],TLRs又称为原型模式识别受体(PRRs),能够识别危险相关分子模式(DAMP)或病原体相关分子模式(PAMPs),从受损的组织或微生物中释放出来[16]。Toll样受体是一类1 型跨膜蛋白,由胞外区、跨膜区和胞内区3个功能区组成。由于其胞内结构域与白细胞介素1(interleukin-1, IL-1) 受体的细胞内区域类似,又被称为Toll/IL-1 受体(Toll/interleukin-1 receptor,TIR) 同源结构域[17],起到调节TLR其下游信号转导通路的作用。
3 TLR4信号通路
作为天然免疫系统的一部分,TLR4通过与脂结合辅助受体MD-2形成复合物来识别细菌细胞表面脂多糖(LPS),并导致细胞内炎症信号的产生[18]。TLR4是唯一一个既可以通过髓样分化因子88 (Myeloid differentiation factor 88, MyD88) 依赖信号通路,又可以通过MyD88非依赖信号通路的Toll样受体[19]。被激活时分别通过诱导活化其下游的IL-1受体相关激酶 (IL-1R associated kinase, I-RAK) 和肿瘤坏死因子受体 (tumor necrosis factor receptor, TNFR) 相关因子6 (TNFR associated factor 6, TRAF-6), 并进一步激活核转录因子 (nuclear transcription factor-kappa B, NF-κB),参与介导细胞凋亡、炎症、氧化应激等多种病理生理过程[20-21]。
4 TLR4的信号通路与青光眼
4.1 TLR4与青光眼的关系 TLR4与青光眼的病理机制有关[22-26],TLR4在人视网膜、虹膜、眼角膜等组织中都有表达。在青光眼患眼中,TLR4表达升高将会导致眼压升高[27]。青光眼是一种由于眼内高压所导致神经退行性疾病,而眼压又受房水循环控制。因此任何引起房水循环阻塞的因素均会形成高眼压而触发视神经损伤机制[28]。
4.2 TLR4的MyD88依赖性通路与青光眼 王乾等[29]研究表明青光眼患者中视野损伤严重的患者其TLR4和 MyD88的表达远高于视野轻度损伤的患者,其过程依照TLR4-MyD88的信号传导。活化的TLR4通过和细胞内TIRAP即含有TIR结构域的接头蛋白,相互作用招募MyD88,激活的MyD88招募下游的TRAK1和IRAK4,活化的IRAK募集并激活下游的TRAF6,此时TRAF6通过形成复合物的形式激活下游的TAK1,TAK1活化下游两条不同途径,即P38MAPK和NF-κB信号级联途径,从而调节各种炎性细胞因子的转录。
4.3 通过TLR4-NF-κB途径影响青光眼视神经损伤 TLR4-NF-κB途径与视神经节细胞的死亡有关[30-31]。正常情况下,神经节细胞会自身凋亡,但是炎症因子的增多会诱导细胞过度凋亡[32]。因此通过TLR4-NF-κB诱导更多的炎症因子产生必然会加重RGC的凋亡。此外,caspase家族在细胞凋亡信号转导中有着重要的作用,而TLR4-NF-κB的通路下游将诱导激活caspase-8信号通路[24],在青光眼中,高眼压的形成会导致TLR4增多,在其通路下激活caspase-8,caspase-8又可以通过两条途径诱导激活IL-β导致神经节细胞死亡,即caspase-1依赖途径(通过调节激活Nod样受体家族的NLRP1和NLRP3炎症反应)和caspase-1非依赖途径。当TLR-4增加,在两条途径共同作用下,IL-β前体转化为IL-β使视神经损伤加重[33]。此外,青光眼患者在缺血的诱导下通过TLR4-NF-κB可使TNF-α、IL-1β、IL-6、Cox-2和MCP-1上调,炎症反应活跃,损伤视神经[34]。TLR4-NF-κB信号通路与青光眼视神经损伤有着正相关的关系基本成立。然而Dvoriantchikova等[35]研究中发现,通过激活TLR4-NF-κB途径存在着保护视网膜缺血的作用,病毒表达活性NF-κB可降低RGCs凋亡(图1)。
4.4 通过TLR4-P38途径影响青光眼视神经损伤 尽管TLR4-NF-κB是TLR4的主要通路,但TLR4-P38对青光眼视神经损伤的影响也仍存在。该途径中P38MAPK的激活使细胞分泌更多的IL-1β,借此可促进分泌Hsp60,Hsp60与TLR4结合,通过MEK 3/6激活P38MAPK,诱导小胶质细胞炎症,这一过程使视网膜的炎症反应不断循环加重,使视神经损伤进一步加深[36-37]。在氧化应激机制中,氧化应激产生的氧化物或者氮化物可通过激活P38MAPK,来激活caspase-3而引起神经节细胞凋亡[38]。
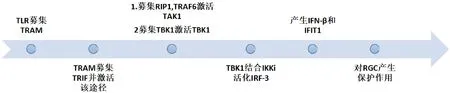
图2 TLR4 通过MyD88非依赖途径对视神经产生保护作用。
4.5 MyD88非依赖途径作用机制 MyD88非依赖途径是通过结合 TRIF的 TLR4实现的,TRIF 活化己结合IKKi的 TBK1,调节IRF-3的活化和核转位,最终使IFN-β 表达;MYD88非依赖途径的TLR4信号在桥梁接头分子 TRAM的作用下,经由 TRIF活化TRAF6也可导致NF-κB活化,进而引起炎症反应。Halder等[39]实验表明在诱导视网膜缺血损伤48h前给予胸腺素原α处理完全防止缺血所致的神经节细胞丧失,双极细胞和光感受器细胞部分存活,其作用原理就是激活TLR4-TRIF-IRF3通路产生保护性基因,抑制TLR4-MyD88-NF-κB途径所产生的损害基因,该实验中TRIF以干扰素β(interferon-β,IFN-β)为代表。除此之外,在该通路的下游还将产生IFIT1和IL1RN等保护性分子。Vartania等[40]实验中证明TLR4-TRIF-IRF3 通路比MyD88-NF-κB和TRIF-NF-κB通路要快和强烈。然而,利用视神经钳夹伤模型研究TRIF在视神经损伤再生中的作用机制,却发现敲除TRIF使RGC存活率增高且视神经再生能力增强[41]。因此该通路对青光眼疾病发展的影响还有待进一步研究(图2)。
5 TLR4与神经胶质细胞相互作用
胶质细胞的激活是多种神经退行性疾病的病理特征,青光眼视神经损伤的发生与神经胶质细胞也有着密不可分的关系。视网膜的炎症改变主要是依赖于其自身的免疫细胞即神经胶质细胞,其中以小胶质细胞为多,M2型的小胶质细胞主要参与炎症损伤,对视神经损伤有着重大影响,此外TLR4主要表达也在神经胶质细胞[42]。正常情况下小胶质细胞可以产生神经营养因子和炎症因子对视神经起营养免疫防御的作用,维持视网膜内环境稳态平衡,当RGCs坏死激发小胶质细胞过度增殖导致内环境紊乱时,破坏血视网膜屏障使神经元坏死加重。神经胶质细胞的增多可诱导TLR4-NF-κB途径[39,43],其通过激活NF-κB通路,分泌 IL-6、TNF-α 等促炎性因子,诱导小胶质细胞的迁移、活化,同时使活化的小胶质细胞更多分化成为 M2 型小胶质细胞[44]。另有报道持续高眼压可引起视神经慢性炎症反应,这一过程就是小胶质细胞激活及激活后TLR-4的信号通路完成[45]。总而言之,TLR4和神经胶质细胞相互作用相互促进,神经胶质细胞在TLR4的刺激下逐渐增多,通过NO和TNF-α诱导RGC凋亡。而TLR4在神经胶质细胞的作用下,产生更多的炎症因子诱导视神经节细胞凋亡(图3)。
6 TLR4作用于小梁网细胞(TM)引起视神经损伤
TLR4除了对视神经节细胞产生直接的损害外还可通过作用于其他组织结构影响视神经细胞的生存。转化生长因子(transforming growth factor, TGF)是多种组织纤维化的主要调节因子,通过细胞外基质增加来引起细胞纤维化。而TLR4已被证实在多个组织中可通过信号通路引起细胞纤维化[46]。在通过TLR4基因突变阻断TGF-β表达的小鼠模型中表明[47],TLR 4通过纤维连接蛋白串扰来控制转化生长因子-β信号的表达,引起小梁网纤维化,导致房水引流阻塞,眼压升高最终使神经节细胞死亡。
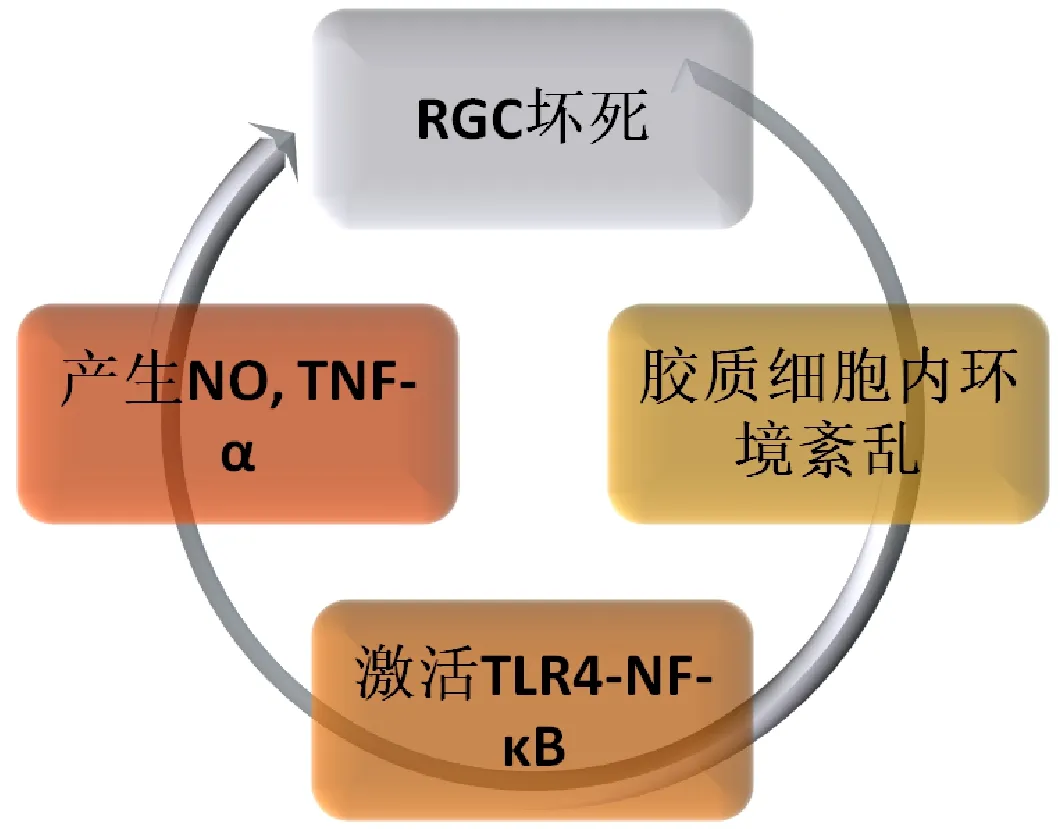
图3 胶质细胞与TLR4共同作用导致视神经损伤的作用机制。
7 展望与讨论
由于青光眼对人们的生活带来了极大的伤害,研究出新的治疗方案刻不容缓,因此对于青光眼发病机制和危险因素的研究成为当今时代的热点,青光眼具有家族聚集性,与年龄、性别、种族都有关系,不同地区青光眼发病的种类也不同,不同基因对疾病易感性不同[48-49],近几年发现,免疫反应对青光眼视神经损伤的影响至关重要,随着科学的发展和进步以及对青光眼视神经损伤机制的研究越加深入,不断揭示了青光眼视神经损伤与TLR4的免疫调节之间的联系,阻断TLR4的信号通路或者抑制神经胶质细胞的激活,可有效减缓青光眼的视神经损伤[50-51],然而TLR4不同信号通路对青光眼具体的作用机制仍存在疑惑,在MyD88依赖性途径在中,TLR4-NF-κB在青光眼视神经的损伤中激活caspase家族加速神经节细胞凋亡,释放多种促炎症因子的物质,炎症反应活跃使视神经损伤。然而Dvoriantchikova等[35]却证实病毒表达活性的TLR4-NF-κB可对视神经节细胞起保护作用,可见该通路的具体影响机制仍存在疑惑。此外TLR4-P38途径对视神经主要起损伤作用,TLR4-TRIF-IRF3 对青光眼视神经损伤却起预防性的保护作用,由此可见,尽管都是TLR4介导的免疫通路其不同的下游激活物对视神经的作用却是相反的,那么在青光眼视神经损害过程中,机体是否存在某些信号物质使保护性通路发挥其最大功效而抑制对视神经起损害作用的途径?小胶质细胞在正常情况下发挥着营养和免疫防御作用,当视神经细胞病理性死亡时才会过多激活其分化增殖通过与TLR4的相互作用,引起内环境紊乱,加重视神经损伤。如何阻断视神经坏死对小胶质细胞的影响,使小胶质细胞仅发挥其神经营养和免疫防御的功能而又不表达过多的TLR4?这些TLR4的矛盾存在是否是因为TLR4的基因多样性,使TLR4发生突变而形成不同的功能。亦或者机体针对TLR4的作用可产生尚未发现的影响因子,对机体各个方面起继发的调节作用。尽管TLR4对青光眼视神经损伤的机制尚未完全开发,但不可否认的是针对TLR4这一作用靶点可对青光眼的治疗起到至关重要的作用。TLR4不同通路之间的平衡如何去调节来治疗青光眼成为进一步需要研究的内容。
猜你喜欢
神经损伤与功能重建(2020年11期)2020-12-01
科学(2020年3期)2020-11-26
中国眼耳鼻喉科杂志(2020年5期)2020-09-30
广东医科大学学报(2020年6期)2020-02-06
中医眼耳鼻喉杂志(2019年3期)2019-04-13
恋爱婚姻家庭(2019年15期)2019-01-27
益寿宝典(2018年11期)2018-01-27
包头医学院学报(2016年2期)2016-06-13
中国中医眼科杂志(2015年1期)2015-12-28
磁共振成像(2015年1期)2015-12-23
