
施奈德结晶状角膜营养不良的分子基础与临床研究进展
2020-05-08 10:58陶俊峰黄玉迪张军林苏振宏解举民
国际眼科杂志 2020年5期
陶俊峰,黄玉迪,张军林,苏振宏, 解举民
施奈德结晶状角膜营养不良(SCCD)是一种稀有的常染色体显性遗传病,其发病部位在眼角膜,伴有结晶状沉淀,双眼发病,家族遗传性,男女患病几率均等。临床研究揭示角膜结晶状混浊化成因是胆固醇、磷脂等脂质在角膜上皮下和基质中异常积累。SCCD的发生与UBIAD1基因突变后脂质代谢异常有关,但是致病的分子机制未知。本文综述了SCCD的发现发展历史、发病分子基础与临床研究,为SCCD的诊疗以及致病分子机制的阐明提供参考。
0 引言
施奈德结晶状角膜营养不良(Schnyder crystalline corneal dystrophy, SCCD;mendelian inheritance in man 121800,MIM 121800) 是一种稀有的常染色体显性遗传病。发病部位在眼部且多为双眼发病。该病最早由法国眼科医师van Went和Wibaut于1924年报道,他们在一个家族中发现8例患者,患者双眼均表现出角膜混浊[1]。瑞士眼科医生Schnyder于1927年、1929年与1939年先后阐明本病的临床表现与遗传特性,故此病被称为SCCD[2-3]。Gillespie和Covelli 1963年报道了1例黑人女性患者,该患者具有典型的SCCD症状,患者父亲、兄长和妹妹同时患有此病,生化检测发现他们均患有高胆固醇血症[3]。有关SCCD疾病的文献,前期主要为病例报道,以及血液生化检测指标,血脂、磷脂及胆固醇含量等。20世纪50年代后,随着分子生物学的发展,检测手段的不断更新,SCCD的临床表现被逐步阐明,该疾病的成因为角膜中脂质异常积累,具体致病分子机制尚不明确。本文系统、全面的综述了SCCD疾病的发现与发展历史,SCCD致病分子基础与临床研究。
1 SCCD的发现与发展
Riegel (1933年),Cavara (1940年),Verryp (1947年),Sysi (1950年), Malbran, Paunessa 和 Vidal (1953年),Glees (1957年)[4],Laurent (1964年),Bisson (1966年),Winkelman (1968年)等分别报道了SCCD病例[5]。1968年,Bron等[6]眼科医生报道了1例新发现的SCCD患者,患者家系第二代中兄妹二人均患有该眼病,先证者为哥哥。Bron团队跟踪监测3a发现该家族中SCCD患者同时伴有高血脂蛋白症,同时对患者进行了细致的检测,发现患者尿液指标正常,但是血检发现患者的胆固醇、甘油三酯和磷脂水平明显高于正常人,在不同的时间内多次检测该家系中患者,结果一致,对SCCD患者的临床表现做了详细的记录,比如结晶状沉淀在角膜的位置、大小、形状、视力水平等,同时他们也综述了从1924年发现该病始到1972年的所有SCCD病例,并附有大部分患者的个人信息与血脂检测结果,他们推测SCCD疾病的发生与血脂异常相关[6]。Michaels[7]于1974年报道了1例SCCD患者,患者20a间眼角膜由斑点状发展成为圆环状针状结晶沉淀,但是该患者的血脂未见异常。Thiel等[8]在IIa型常染色体显性高脂蛋白血症家系中,发现了1例19岁男性患者同时患有SCCD疾病,而且从1964年~1976年间角膜逐渐混浊化。Burns等[9]运用14C标记的胆固醇示踪,发现全层角膜移植后,角膜中胆固醇含量比血清中高,证明角膜是胆固醇吸收和存储的重要活性部位。Ingraham[10]发现SCCD患者随时间延长角膜混浊化加重,纠正了之前研究SCCD在患病后不会继续发展的错误,同时综述了前人的研究结果,认为只有部分SCCD患者会伴随高血脂症与高胆固醇血症,而且这两项指标并不能用于确诊SCCD疾病。Martin 等[11]使用定量生化检测方法测得SCCD患者角膜的脂质异常积累,主要包含磷脂、游离胆固醇和胆固醇酯,较之对照角膜,患者角膜中磷脂含量为23.6mg/gvs4.05mg/g,游离胆固醇 6.99mg/gvs0.52mg/g,胆固醇酯3.16mg/gvs0.26mg/g,因此Ingraham[10]推测SCCD的发病机制为角膜中脂质代谢异常。Weiss[12]为了检测SCCD患者角膜结晶化出现比率,自1987年始共收集33例SCCD患者并对患者做了全面的眼科检查发现只有17例(51.5%)患者角膜中出现结晶状沉积,而该疾病SCCD的称法对眼科医生而言是一种错误引导,所以他们建议将该疾病重新命名为Schnyder corneal dystrophy (SCD)[12]。SCCD患者的临床表现包括角膜混浊化、角膜老年环和膝关节外翻等,Battisti等[13]报道了1例SCCD患者血脂正常,但是皮肤和体外培养的成纤维细胞中出现脂质异常积累,可能由细胞内胆固醇代谢异常导致。SCCD是一种典型的家族遗传病,遵循孟德尔遗传定律,其特点为常染色体显性遗传,推测该疾病的病因学基础为基因缺失或突变。随着分子生物学的飞速发展,很多疾病的分子基础被阐明,探寻SCCD疾病的分子基础也成为科学问题之一。
2 SCCD的分子基础
Takeuchi等[14]发现了一个新的人类cDNA分子,将其命名为B120。使用荧光原位杂交方法检测到该基因定位于1号染色体p35~36.1,将该基因片段导入表达载体后,转入Cos1,C3H/10T1/2和NIH/3T3细胞后,检测到细胞质中出现了球形小泡,使用电子显微镜和免疫组化染色的方法鉴定该小泡为脂肪液滴。该结果证明B120基因与细胞内的脂质代谢相关,在不同细胞(包括成纤维细胞)中过表达B120基因,可以导致细胞内脂质沉积,出现脂肪小泡。眼角膜主要由成纤维细胞组成,所以B120基因的过表达可能与SCCD疾病产生相关[14],该研究与之前另一篇关于SCCD疾病发生可能是特定基因遗传紊乱造成的结论不谋而合[15]。随着分子生物学和分子遗传学的发展,新技术和方法的不断涌现,疾病发生与基因的关系被不断阐明。Auw-Hadrich等[16]利用分子遗传学的方法将眼角膜营养不良疾病与基因关联起来,他们将角膜营养不良疾病做了区分:BIGH 3基因突变可以导致角膜前膜营养不良,如Ⅰ型和Ⅱ型颗粒状角膜营养不良,Ⅰ型和ⅢA型晶格状角膜营养不良;9号染色体q34突变可以导致Ⅱ型晶格状角膜营养不良;16号染色体q22突变可以导致角膜斑点状营养不良;1号染色体p36突变可以导致 SCCD;20号染色体p11.2~q11.2突变可以导致施列丁后部多形性角膜营养不良。该文章综述了多种角膜营养不良疾病的发生与基因的关系,为SCCD疾病病因学的阐明奠定了分子基础。Riebeling等[17]报道了一个SCCD家系,该家系中66岁的母亲患有SCCD、Ⅳ型高脂蛋白血症和高胆固醇血症,她的一个儿子患有SCCD和高胆固醇血症同时伴有LDL-胆固醇含量升高。微卫星分析发现患病儿子与非患病儿子在1号染色体p34.1~p36,D1S228标签附近含有不同的等位基因。因此推测SCCD的发生与D1S228标签附近基因异常有关。早在1995年Amanda等[18]分析了马萨诸塞州中部两个瑞典-芬兰后人家系,分析了基因组中300个微卫星标记,90%以上的基因组序列被排除,推测SCCD的基座位于1号染色体p34.1~p36之间,通过单体型分析最终推测SCCD的基座位于D1S2663和D1S228两个标签之间16厘摩的间隔中。时隔8a后,同一科研团队Theendakara等[19]又收集了来自芬兰、土耳其、德国和美国的13个家系并做了精细分析,揭示SCCD基座位于D1S244和D1S3153两个标签之间1.58M碱基对内,该结果进一步缩小了SCCD基座在基因组中的定位,为筛选SCCD疾病的致病基因奠定了基础。Aldave等[20]筛选了两个家系SCCD基座区15个候选基因 (CORT,CLSTN1,CTNNBIP1,DFFA,ENO1,GPR157,H6PD,KIF1B,LOC440559,LZIC,MGC4399,PEX14,PGD,PIK3CD,SSB1),印证了之前报道中的17个单核苷酸多态性(SNP)位点,同时也发现了一批新的SNP位点,GPR157基因[c.795 C>T (Arg 218 Leu); c.811 C>T (Ala 223 Val)],MGC4399基因[c.1024 G>C (Leu 277 Leu)],和H6PD基因[c.754 A>C (Asp 151 Ala)]。但是Aldave等[20]分析的15个候选基因并不是SCCD的致病基因,这已经排除了一半的候选基因,在剩余的基因中有2/3功能已知,本项工作为进一步揭示SCCD致病分子基础做了重要的指导。直至2007年,SCCD致病基因被发现,距离第一次报道该疾病已经过了80多年。 SCCD的病因是角膜局部脂质异常积累,在多数病例中发现血脂代谢紊乱。Orr等[21]在一个几代人的大家系中利用精细定位分析推测1.3Mbp间隔中候选基因UBIAD1的突变是引起SCCD的分子基础,并且在其他几个SCCD家系中发现了UBIAD1基因突变,UBIAD1是一个异戊烯转移酶,与载脂蛋白E相互作用,参与细胞内的胆固醇代谢,其具体分子机制未知。Weiss等[22]分析了6个SCCD家系中患者的DNA样本,排除了FRAP1、ANGPTL7两个基因,发现了UBIAD1基因突变与SCCD相关,在其中的5个家系中发现了第102位氨基酸由天冬氨酰突变为丝氨酸(N102S),该结果与Orr报道的一个家系中UBIAD1突变一致,另外一个家系中第177位氨基酸由甘氨酸突变为精氨酸(G177R)。UBIAD1基因编码一个异戊烯转移酶,参与体内的胆固醇合成,SCCD的产生可能与角膜中胆固醇的异常积累有关。根据研究结果推测SCCD的发病原因为UBIAD1基因突变,异戊烯转移酶功能受损,角膜中胆固醇等脂质代谢异常,导致脂质在角膜中积累、沉淀或结晶化。SCCD的分子基础经过多位科学家多年的努力终于被阐明,这一发现为SCCD疾病治疗与药物研发提供了分子靶标与参考。

图1 患者为14岁男性,角膜上皮下局部弧状结晶。
图2 患者为28岁女性,角膜上皮下近环状结晶。

图3 患者为37岁男性,角膜中部薄雾化出现。
图4 患者为40岁男性,角膜中部圆盘状混浊化,弧状类脂沉积。
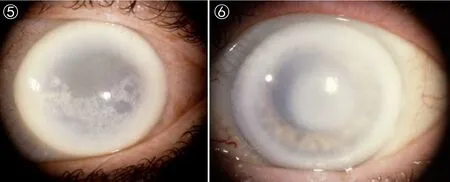
图5 患者为63岁女性,角膜上皮下结晶化,角膜呈弥散型薄雾化,有弧状类脂。
图6 患者为72岁女性,角膜混浊化严重,角膜中央外周薄雾化,弧状类脂积累多。
3 SCCD的临床表现与治疗
3.1 SCCD的临床表现 SCCD是一种稀有的常染色体显性遗传病,其特点为连续性双眼角膜混浊化,成因为胆固醇、磷脂等在角膜中的异常积累。大部分SCCD患者血脂、脂蛋白和胆固醇含量异常,但是角膜逐渐混浊化和结晶化与血脂并没有直接关系[23]。临床检测发现约有54% SCCD患者角膜中有结晶状沉淀,而另外46%的患者并不伴随角膜结晶状沉淀出现,所以国际命名委员会应Weiss等[24-26]眼科学家的要求将SCCD更名为SCD(本文为避免混淆,统一使用SCCD)。化学方法检测SCCD患者手术移除的眼角膜中胆固醇含量增加了10倍,磷脂含量增加了5倍,HDL中的载脂蛋白异常积累[27]。
SCCD的发病年龄可以划分为3个阶段:(1) 26岁及以下;(2) 26~39岁;(3) 40岁及以上。不同参考文献对年龄划分存在差异,还有一种划分方式时间节点为23和38岁,目前发现最小的SCCD患者为17月龄[22],最大为81岁,平均发病年龄为38.8±20.4岁[23, 25]。
SCCD的临床表型为:(1)早期(≤26岁):在角膜基质上皮下中央位置出现结晶状沉淀(图1、2)[23]。(2)中期(>26~<39岁):结晶状沉积不断积累,并出现薄雾化(图3、4)[23]。(3)晚期(≥39岁): 随年龄的增长,结晶化程度不断加重,导致整个角膜混浊化(图5、6)[23]。从角膜中结晶状沉淀的积累,推知该疾病随年龄增长持续性发展,导致视力逐渐缺失,最终失明。约有4%的SCCD患者中同时伴随膝外翻或叉型腿等表型[23, 28-29]。
3.2 SCCD的治疗方法 SCCD患者的视力随着年龄增长逐渐丧失,40岁之前视力可以通过矫正达正常视力水平。40岁以后角膜混浊化加重,视力丧失程度加剧。SCCD患者主要通过穿透性角膜移植(penetrating keratoplasty, PKP)和光治疗性角膜切削术(phototherapeutic keratectomy, PTK)方法来恢复视力[23, 30],约有54%的患者会在50岁后,约有77%的患者在70岁以后借助穿透性角膜移植手术恢复视力[30]。
4 总结与展望
SCCD是一种稀有的常染色体显性遗传病,在男女中发病几率均等,随年龄增长角膜混浊化加剧最后导致视力丧失,其原因为1号染色体短臂1p36区带UBIAD1基因突变,导致角膜中胆固醇、磷脂等在角膜中异常积累[30],目前在多个国家发现了该病病例[3, 15, 20-21, 29, 31-35]。国内眼科学者也报道过该病例,Jing等[29]报道在河南省发现了1例29岁女性SCCD患者,该患者同时伴有膝关节外翻。复旦大学附属眼耳鼻喉科医院张朝然团队也报道过一个患病家系,该家系中共有5例患者,其中有1例患者在临床检查中未发现角膜中结晶状沉淀[36]。日本学者报道UBIAD1是人体内的一种4-甲基萘醌合成酶[37],该文章拓展了UBIAD1基因的功能,为进一步揭示该基因在体内参与的生理生化过程以及导致SCCD疾病的分子机制提供了参考。截止目前,有关SCCD的研究报道不足150篇,该疾病致病基因UBIAD1的功能研究正在逐步开展,为最终阐明SCCD的发病机制夯实基础。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
科学之谜(2019年3期)2019-03-28
吉林林业科技(2018年6期)2018-11-21
中成药(2018年9期)2018-10-09
科学之谜(2018年8期)2018-09-29
中成药(2018年1期)2018-02-02
中成药(2017年4期)2017-05-17
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
听力学及言语疾病杂志(2015年5期)2015-12-24
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
