
功能性乙基纤维素-松香复合膜的合成与性能表征
2020-05-06 11:31丘雨玲郭晓亮卢传巍王春鹏王基夫储富祥
林产化学与工业 2020年2期
丘雨玲, 郭晓亮, 卢传巍, 王春鹏, 王基夫*, 储富祥
(1.中国林业科学研究院 林产化学工业研究所;生物质化学利用国家工程实验室;国家林业和草原局 林产化学工程重点实验室;江苏省生物质能源与材料重点实验室,江苏 南京 210042; 2.南京林业大学 江苏省林业资源高效加工利用协同创新中心,江苏 南京 210037)
化石资源的大量开发一方面促进国家经济高速发展和满足人们日常生活需要,另一方面已造成日益严重的环境问题,将天然可再生的生物质取代化石资源用于制备高分子材料成为了研究热点[1-5]。纤维素作为自然界来源最广泛的天然资源[6],通过利用纤维素分子上的羟基,已开发出大量纤维素衍生物[7-8]。为了进一步拓展纤维素的应用领域,许玉芝[9]将月桂酸与纤维素进行酯化反应合成具有内塑化性能的纤维素酯类衍生物。Liu等[10]通过原子转移自由基聚合(ATRP)反应将松香和甲基丙烯酸月桂酯共聚物接枝到纤维素分子侧链上,制备了可持续使用的新型热塑性弹性体(TPE),该研究可通过改变侧链中松香与甲基丙烯酸月桂酯的比例来调控弹性体的机械性能。Goldmann等[11]采用可逆加成-断裂链转移(RAFT)聚合法和DA环加成法相结合,在(T≈20 ℃)快速和模块化条件下,将聚丙烯酸异冰片酯接枝到固体纤维素上。乙基纤维素(EC)是一种非常重要的商品化纤维素醚类衍生物,主要用作片剂黏合剂和薄膜包衣材料等[12-14],然而由于纤维素的刚性分子结构以及羟基的氢键作用力,EC作为膜材料使用时,其韧性不足,在室温(22 ℃)条件下呈现出明显的脆性,实际应用中常需对EC进行共聚、接枝等改性[15-17]。松香是由多种树脂酸组成的混合物,其三环二萜骨架结构上含有碳碳不饱和键及羧基,有利于进行加成和酯化改性[18]。基于EC和松香结构上的特点,本课题组前期研究[19]已通过酯化反应将松香树脂酸接枝到EC分子侧链上,构筑出一类侧基含有三元环状分子结构的新的纤维素衍生物,并实现了松香树脂酸分子的刚性、疏水性及紫外光吸收性与纤维素复合。马来海松酸(MPA)是一种非常重要的松香衍生物,相对于松香树脂酸,马来海松酸在分子结构上含有酸酐和羧基官能团,可用作环氧固化剂,将MPA与纤维素进行复合的研究尚未见报道。本研究以马来海松酸为原料,酰氯化后与EC酯化,合成了乙基纤维素马来海松酸酯(EC-g-MPA),并以环氧大豆油(ESO)作增塑剂,制备了环氧大豆油松香-纤维素基复合物膜(EC-g-MPA-ESO),探讨了复合膜的力学性能、紫外吸收性能及热稳定性等。
1 实 验
1.1 材料与仪器
乙基纤维素(EC),在甲苯中的黏度270~330 mPa·s,草酰氯(纯度98%)、 4-二甲氨基吡啶(DMAP, 纯度99%),阿拉丁试剂公司;四氢呋喃(THF,纯度99%)、甲醇、 N,N-二甲基甲酰胺(DMF)、二氯甲烷(DCM),均为分析纯。马来海松酸(MPA,纯度>99%),自制[20]。
Nicolet iS10傅里叶红外光谱(FT-IR)仪;Bruker DRX500型核磁共振(NMR)仪,瑞士Bruker公司;UV240紫外-可见分光光度计(UV-Vis),日本Shimadzu;NETZSCH STA409PC型热重(TG/DTG)分析仪;Perkin Elmer Diamond差示扫描量热分析(DSC)仪;CMT7504万能试验机。
1.2 乙基纤维素马来海松酸酯的制备
为了进一步实现EC的功能化,在本研究中将利用EC剩余的羟基官能团,通过酯化反应引入MPA分子,赋予EC新的功能。MPA是一种具有三元环的分子,其羧基官能团位于MPA分子的叔碳位置,因而羧基官能团具有较大的位阻效应。因此,为了提高MPA分子的反应活性,在与EC进行酯化反应之前,先将MPA的羧基转化成酰氯基官能团,然后再与EC进行酯化反应,制备出乙基纤维素马来海松酸酯(EC-g-MPA),合成路线如图1所示。
取26 g(0.065 mol)MPA溶于40 mL THF中,在0 ℃条件下滴入7.2 mL(0.084 5 mol,10.7 g)草酰氯,并滴加0.35 mL DMF,50 ℃反应2 h后,去除溶剂得到中间产物马来海松酸酰氯(MPACl)。将MPACl溶解在THF中,滴加到含有EC(12.3 g,0.05 mol)和DMAP(6.5 g)的 THF溶液中,将反应体系升温至50 ℃反应1 h后,冷却至室温,离心取上层清液,倒入过量蒸馏水中,过滤,先后经碱水洗、酸水洗和去离子水洗后,再将固体产物溶解在甲醇中,加入大量去离子水使固体产物析出,收集固体,在60 ℃ 真空干燥24 h,得到EC-g-MPA。
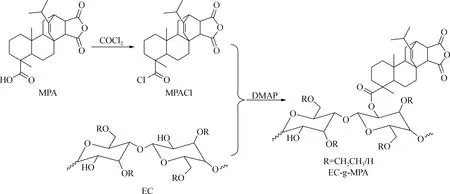
图1 EC-g-MPA的合成路线Fig.1 Synthesis of EC-g-MPA
1.3 EC-g-MPA-ESO膜的制备
将MPA分子结构引入EC中,由于MPA分子的刚性作用,EC-g-MPA膜材料的韧性有待提高。因此,为了提高EC-g-MPA膜材料的力学性能,选用可再生的环氧大豆油(ESO)作为改性剂,探讨了不同的ESO加入量对膜材料力学性能的影响。以ESO为增塑剂,按照ESO用量(以EC-g-MPA质量计)为0%、5%、10%、15%、20%、25%和30%与EC-g-MPA混合并溶于二氯甲烷,倒入聚四氟乙烯模具中,25~30 ℃ 条件下静置48 h,并经过150 ℃热压后得到环氧大豆油松香-纤维素基聚合物(EC-g-MPA-ESO)膜。
1.4 分析与表征
1.4.1FT-IR分析 采用Nicolet iS10傅里叶红外光谱仪测试样品的红外吸收特征情况。
1.4.21H NMR分析 采用Bruker DRX500型核磁共振仪,氘代氯仿作为溶剂,进行样品分析。
1.4.3UV-Vis分析 使用紫外-可见分光光度计测试聚合物膜的紫外吸收性能。
1.4.4热力学性能分析 使用NETZSCH STA409PC型热重分析仪测定样品的热失重性能,设置氮气流速为40 mL/min,升温速率为15 ℃/min,升温范围为40~800 ℃;采用差示扫描量热分析(DSC)仪测试样品的玻璃化转变温度(Tg)。
1.4.5力学性能分析 使用CMT7504万能试验机测试膜样品的力学性能,测试温度为22 ℃,测试拉伸速度为50 mm/min,传感器载荷为1 000 N。
2 结果与分析
2.1 EC-g-MPA的结构分析
2.1.1FT-IR分析 马来海松酸(MPA)、中间产物马来海松酸酰氯(MPACl)和乙基纤维素马来海松酸酯(EC-g-MPA)的红外光谱见图2。MPA在1842和1772 cm-1处有明显的酸酐特征官能团吸收峰,1692 cm-1为羧基的特征吸收峰。当MPA进行酰氯化反应之后,在1692 cm-1处的羧基特征吸收峰消失,产生酰氯特征吸收峰与1772 cm-1处的酸酐特征官能团吸收峰重叠。经酯化反应后,在1725 cm-1处新生成了酯基特征峰,同时在1842和1772 cm-1处仍明显观察到酸酐特征官能团吸收峰,以上结果表明,MPA已成功接枝到EC上,并将酸酐官能团引入EC中。
2.1.21H NMR分析 EC-g-MPA的1H NMR谱图见图3。EC中甲基上质子的化学位移在δ1.2,MPA分子的引入,其亚甲基上质子的化学位移在δ1.2~2.0。由图3可看到EC-g-MPA上MPA的不饱和双键的质子峰(δ5.6),证实MPA已成功接枝到EC分子上。
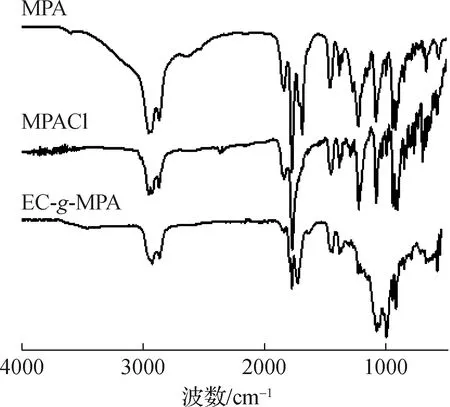
图2 不同样品的FT-IR谱图
Fig.2 FT-IR spectra of different samples
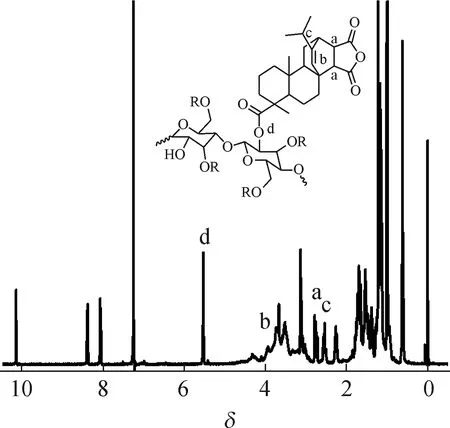
图3 EC-g-MPA的1H NMR谱图
Fig.31H NMR spectrum of EC-g-MPA
2.2 EC-g-MPA-ESO膜的结构和性能
2.2.1UV-Vis分析 通过将MPA接枝到EC骨架上制备出EC-g-MPA,并研究其成膜性,然后进行UV/Vis吸收测试,结果如图4所示。
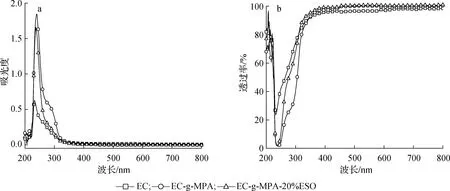
图4 不同样品的紫外吸收(a)和透射(b)光谱图Fig.4 UV-visible absorption(a) and transmittance(b) of different samples
由图4(a)和(b)可见,相对于EC,EC-g-MPA在200~300 nm存在UV吸收和过滤,这表明EC-g-MPA与MPA一样,具有一定的紫外光吸收和过滤性能,同时也进一步证实了MPA成功接枝到EC骨架。EC-g-MPA-20%ESO的紫外吸收和透过率曲线在200~250 nm区间大致与EC-g-MPA重合,而在250~300 nm区间,EC-g-MPA-20%ESO紫外吸收强度明显大于EC-g-MPA,相对应的EC-g-MPA-20%ESO的紫外透过率明显低于EC-g-MPA。松香的主要成分树脂酸的三环菲结构中含有多个不饱和双键,其分子在聚合物中能形成π-π堆积,从而具有一定的紫外吸收和过滤特性,该现象已在本课题组前期的研究工作中得到证实[16,21]。将MPA与EC进行接枝反应后,聚合物分子中仍然含有不饱和双键,使得EC-g-MPA薄膜具有紫外吸收和过滤的性能。将ESO引入EC-g-MPA中,由于ESO分子中的环氧基与EC-g-MPA分子中的酸酐基团进行开环反应,使EC-g-MPA-20%ESO的紫外吸收强于EC-g-MPA,相对应的使EC-g-MPA-20%ESO的紫外透过率强于EC-g-MPA。
2.2.2FT-IR分析 为探明ESO对EC-g-MPA增韧及EC-g-MPA的Tg下降的原因,采用红外光谱对EC-g-MPA-20%ESO的结构进行表征,如图5(a)所示。相比于EC-g-MPA的红外光谱图, EC-g-MPA-20%ESO在1842和1772 cm-1处的酸酐特征官能团吸收峰消失,同时在1730 cm-1处的酯基峰得到增强。另外,在3400 cm-1处的也有ESO开环反应后生成的羟基吸收振动峰,这表明ESO的环氧基与EC-g-MPA的酸酐官能团进行酯化反应,实现了ESO与EC-g-MPA的共价键连接,从达到了EC-g-MPA内塑化的目的。
2.2.3热性能分析
2.2.3.1DSC分析 从图5(b)可以看出,EC-g-MPA的玻璃化转变温度(Tg)为90 ℃,相比EC的Tg(102.5 ℃)略有下降,但对于膜材料,特别是具有柔韧性的膜材料,Tg仍相对较高。因此,加入ESO对EC-g-MPA进行改性和增塑,获得的EC-g-MPA-20%ESO的Tg为15 ℃。
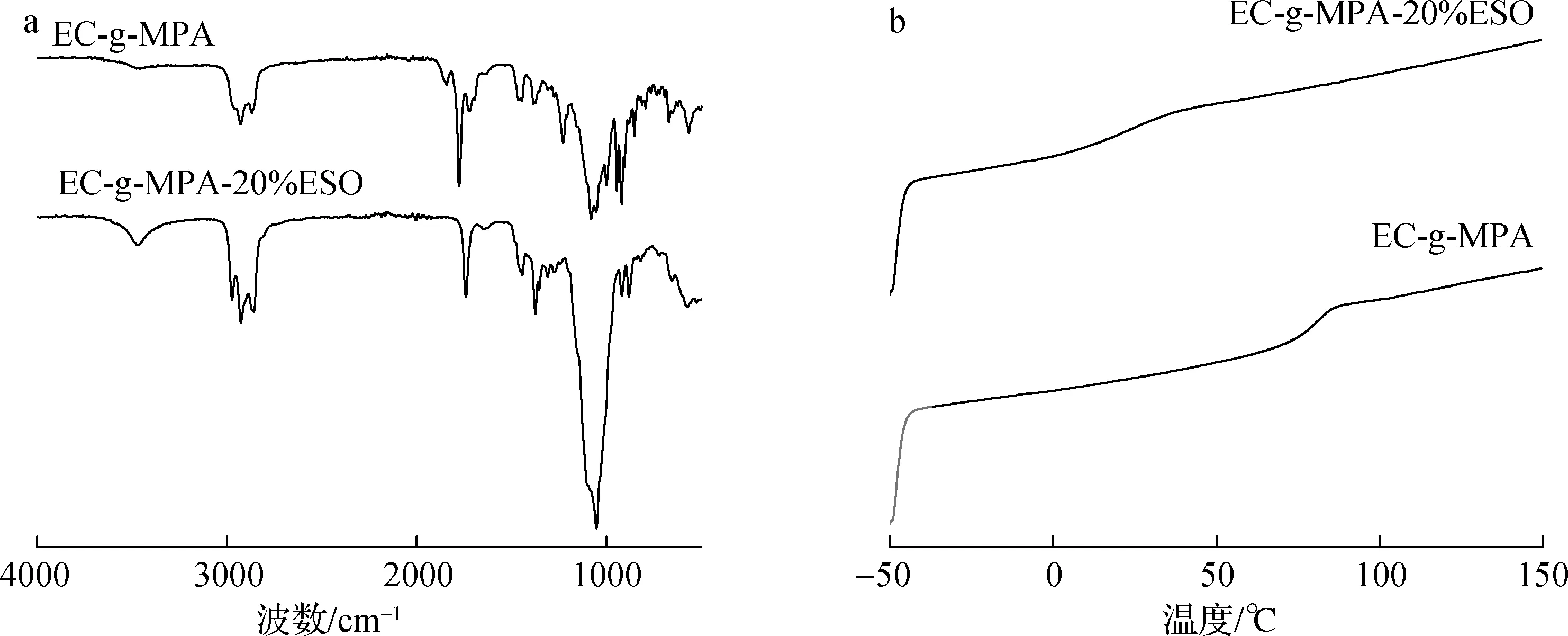
图5 不同样品的FT-IR图(a)和DSC曲线(b)Fig.5 FT-IR spectra(a) and DSC curves(b) of different samples
2.2.3.2TG/DTG分析 图6为EC-g-MPA及其ESO膜的TG和DTG曲线。
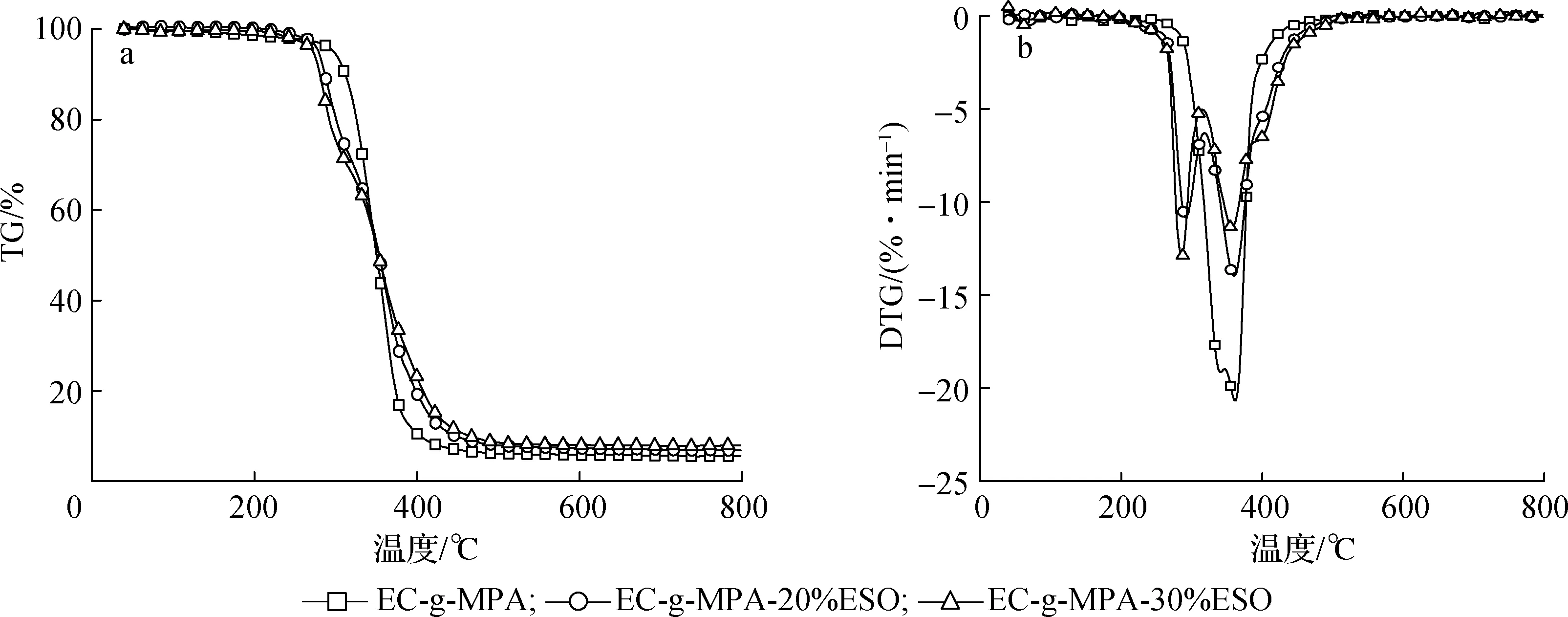
图6 不同样品的TG(a)和DTG(b)曲线Fig.6 TG(a) and DTG(b) curves of different samples
从图6(a)可以发现EC-g-MPA的热分解温度开始于316 ℃左右,而EC-g-MPA-20%ESO和EC-g-MPA-30%ESO的热解温度都始于280 ℃左右。结合图6(b)可以发现EC-g-MPA-20%ESO和EC-g-MPA-30%ESO的TG和DTG曲线趋于一致,这说明两者的组成成分大致相同,相比于EC-g-MPA的TG和DTG,EC-g-MPA-20%ESO和EC-g-MPA-30%ESO的TG和DTG曲线明显分为3个阶段,从280~300 ℃左右EC-g-MPA-20%ESO和EC-g-MPA-30%ESO分解速率比EC-g-MPA大,这可能是由于复合膜中未参与反应的小分子挥发或者分解造成的现象。从300~400 ℃阶段3个样品DTG曲线的峰形状大致相似,这可能是EC-g-MPA的主要分解阶段。而400 ℃以后直至分解完全阶段, EC-g-MPA-20%ESO和EC-g-MPA-30%ESO分解速率大于EC-g-MPA,这说明复合膜中产生了部分比EC-g-MPA更难以分解的组分,这可能是ESO与EC-g-MPA发生反应生成了更复杂的物质,进一步佐证了ESO分子成功接枝到EC-g-MPA链段中。
2.2.4力学性能分析 不同的ESO用量可以对膜材料机械性能进行调控,基于性能比较的目的,同样采用环氧大豆油对乙基纤维素松香基聚合物进行增塑和成膜实验,实验结果见表1。
结合表1和图7可以发现,在没有加入ESO进行内增塑之前,EC-g-MPA膜拉伸强度只有2.08 MPa;当ESO用量从5%增加到15%时,其拉伸强度从5.52 MPa增加到7.54 MPa。当ESO用量达到20%时,其拉伸强度达到最大值12.07 MPa,并且其应变明显增加;当ESO用量继续增加到30%时,其拉伸强度下降至7.01 MPa。总体来看,当ESO用量为20%时膜材料的力学性能最优。图8为EC-g-MPA-20%ESO的循环拉伸应力-应变曲线,由图可以看出,复合膜具有优异的弹性体性能,且弹性恢复系数随着伸长率的增加而增加,当伸长率达到80%时,其弹性恢复系数可以达到54.6%。
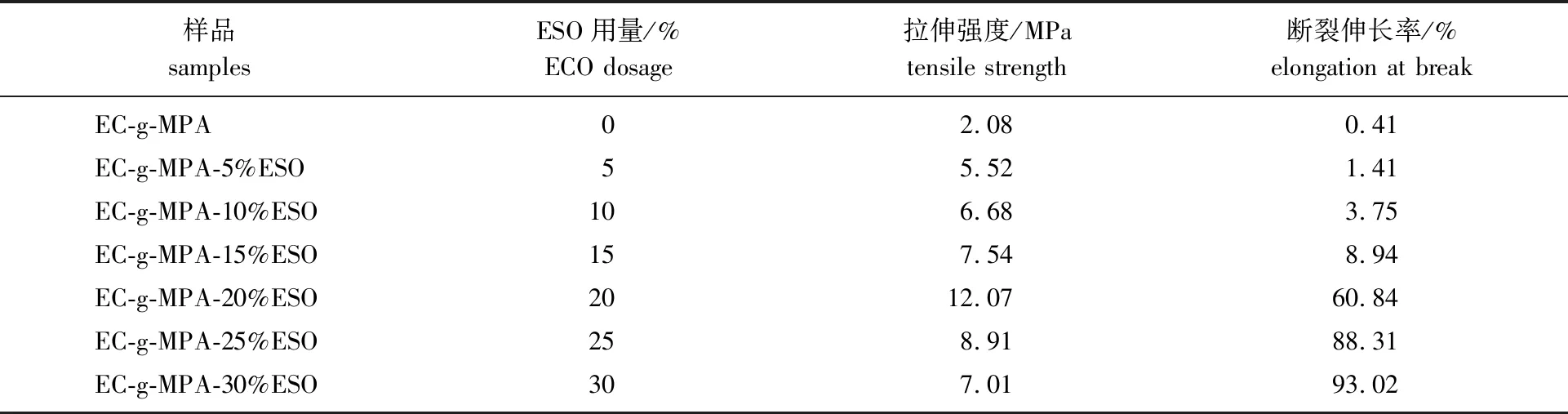
表1 EC-g-MPA-ESO的机械性能结果Table 1 Mechanical properties of EC-g-MPA-ESO
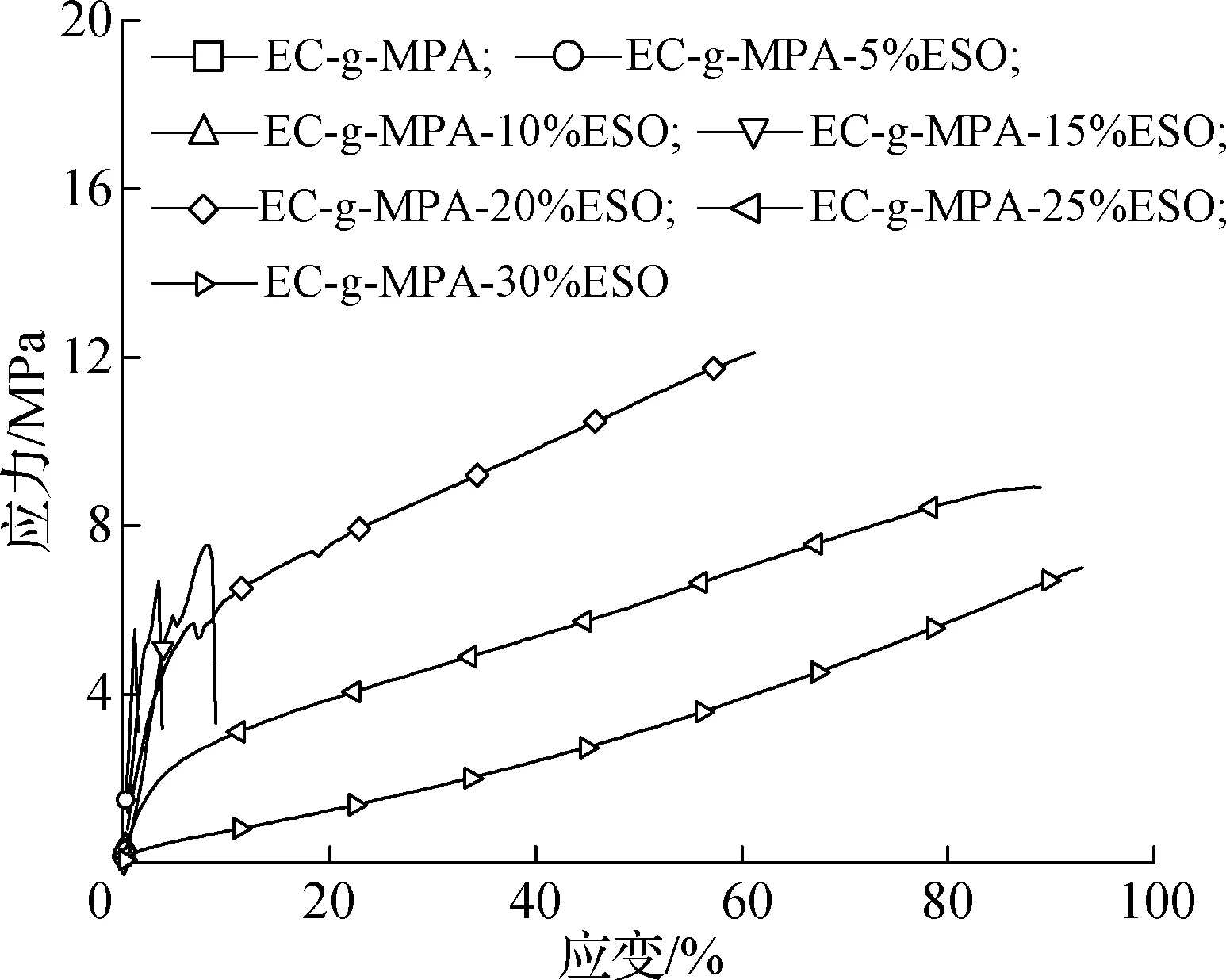
图7 EC-g-MPA-ESO拉伸应力-应变曲线
Fig.7 Stress-strain curves of EC-g-MPA-ESO
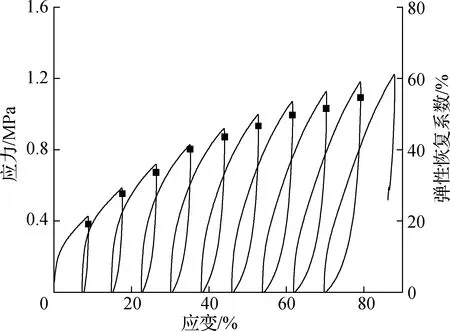
图8 EC-g-MPA-20%ESO的循环拉伸应力-应变曲线
Fig.8 Cyclic stress-strain curves of EC-g-MPA-20%ESO
3 结 论
3.1马来海松酸(MPA)经过酰氯化后,可以成功接枝到乙基纤维素(EC)分子上,合成乙基纤维素马来海松酸酯(EC-g-MPA),通过FT-IR、1H NMR和UV-Vis证实马来海松酸已成功接枝到乙基纤维素分子上。
3.2采用环氧大豆油(ESO)对EC-g-MPA进行改性和内增塑,获得EC-g-MPA-ESO复合膜。相比于EC和EC-g-MPA,复合膜具有较低的玻璃化转变温度和较好的韧性。
3.3当ESO用量达到20%时,EC-g-MPA-ESO拉伸强度达到最大值12.07 MPa,力学性能最佳;循环拉伸实验证实EC-g-MPA-ESO弹性恢复系数随着伸长率的增加而增加,当伸长率达到80%时,其弹性恢复系数可以达到54.6%,表明EC-g-MPA-ESO具有优异的回弹性,可作为热塑性弹性体。
猜你喜欢
宝藏(2021年4期)2021-05-27
林产化学与工业(2021年2期)2021-05-11
紫禁城(2020年5期)2021-01-07
林产化学与工业(2020年5期)2020-11-04
林产化学与工业(2020年3期)2020-06-30
小学生学习指导(低年级)(2017年11期)2017-10-23
中学生(2017年13期)2017-06-15
数学大王·中高年级(2016年10期)2016-09-10
中国塑料(2015年5期)2015-10-14
传奇故事(破茧成蝶)(2015年1期)2015-02-28
