
无肌病性皮肌炎一例
2020-04-30 01:21周桂芝杨宝琦
中国麻风皮肤病杂志 2020年4期
曹 珊 周桂芝 杨宝琦
1济南大学山东省医学科学院医学与生命科学学院,济南,250062;2山东第一医科大学附属皮肤病医院(山东省皮肤病医院),山东省皮肤病性病防治研究所,济南,250022
临床资料患者,女,21岁。因面部、躯干、四肢皮肤红斑、丘疹伴脱发6年,于2019年3月20日来我院就诊。6年前患者日晒后双侧眼睑、两侧面颊及鼻背部皮肤起红斑、丘疹,后蔓延至颈部V型区、后背及双上肢近端、双侧股部,渐散在分布全身,病程中无发热、关节疼痛、雷诺症、吞咽及呼吸困难等症状,6年前曾在当地医院诊断为“日光性皮炎、湿疹”,住院治疗,具体诊疗不详;5年前另外一家医院诊断为“皮肌炎”,口服糖皮质激素治疗,剂量不详,部分皮损消退,后因拟妊娠停药。既往体健。1年半前顺产1女孩,体健。系统查体:浅表淋巴结未触及肿大。四肢无压痛,四肢肌力正常。皮肤科查体:皮损弥漫性分布于躯干、四肢及面颈部,基本损害为红斑,面部尤以双侧眼睑见明显水肿性紫红斑,其余皮损处见弥漫性对称性暗紫红色斑,部分融合成片,境界不清,双手可见Gottron丘疹(图1)。毛发轻度稀疏,指趾甲未见明显异常。实验室检查:尿酸443 mmol/L,肌酸激酶同工酶29 U/L,α-羟丁酸脱氢酶194 U/L,谷草转氨酶、肌酸激酶、乳酸脱氢酶均正常;免疫球蛋白IgE 1783 kU/L,免疫球蛋白IgG 10.82 g/L、IgA 1.16 g/L、IgM 0.66 g/L;补体C3 1.12 g/L、C4 0.28 g/L;血常规、肝肾功能、尿常规、血沉、血卟啉、尿卟啉均正常,抗核抗体(ANA)实验1∶100阳性、均质性,抗双链DNA及抗盐水可提取物抗原(ENA)、抗Sm抗体未见异常。肌炎相关抗体中抗T1FI-γ抗体阳性,抗MDA5抗体、抗Jo-1抗体、抗PL-7抗体、抗PL-12抗体、抗MI-2α抗体、抗MI-2β抗体、抗NXP2抗体、抗SAE1抗体、抗RO-52抗体、抗PM-SCL75抗体、抗PM-SCL100抗体及抗KU抗体均阴性,肿瘤标记物中甲胎蛋白、癌胚抗原、糖链抗原19-9、糖链抗原125及糖类抗原15-3均未见异常。左上肢皮损活检组织病理示:表皮轻度角化过度,基底层增厚,真皮浅层水肿,血管周少许单一核细胞浸润(图2a)。直接免疫荧光(DIF):基底膜带IgG、C3、IgM阳性带状沉积,IgA弱阳性带状沉积(图2b)。胸部正侧位数字化摄影和腹部彩超未见异常。双肺CT检查未见明显异常。诊断:无肌病性皮肌炎。治疗:给予羟氯喹0.2 g,每日2次,雷公藤2片,每日3次,薄芝片0.48 g,每日2次,外用多磺酸粘多糖乳膏及丁酸氢化可的松乳膏。
1个月后复诊,系统查体及皮肤科查体基本同前,实验室检查:肌酸激酶同工酶27 U/L,其余正常。给予甲氨蝶呤15 mg,每周1次,其余治疗同前。再次治疗1个月后复诊,系统查体同前,皮肤科查体见皮损颜色仅轻度变淡。实验室检查:尿蛋白+-,尿比重1.031(正常1.01~1.03),尿胆原2+,肌酸激酶同工酶32 U/L,免疫球蛋白IgE 1426 U/L,其余正常。给予醋酸泼尼松40 mg/d,碳酸钙D3片1片/d,其余治疗同前。泼尼松联合甲氨蝶呤治疗2个月后复诊原有皮损颜色变淡,眼眶周紫红色水肿性斑片消失,面部皮损基本消退,后背及双臂、双下肢皮肤异色症减轻,双手Gottron丘疹大部分消退(图3),未见新发皮疹。实验室检查:肌酶、肝肾功及血常规均正常。醋酸泼尼松逐渐减量至20 mg/d,其余药物不变。目前病情平稳,仍在继续随访中。
讨论1979年Pearson提出“无肌病性皮肌炎”概念,并认为无肌病性皮肌炎是皮肌炎的一种临床亚型[1]。无肌病性皮肌炎(amyopahic dermatomyositis,ADM)是一组具有经活检证实的特征性皮肌炎的皮疹表现,但同时不具有近端肌无力和血肌酶谱的升高,且病程超过6个月以上[2],属于临床无肌病性皮肌炎(clinical amyopathic dermatomyositis,CADM)的一种类型。ADM发病机制及发病率尚不明确,Chen等[3]报道CADM占皮肌炎的8.8%(29/331)。国外学者发现特发性炎性肌病患者体内存在一个抗体,该抗体识别的靶抗原为转录中介因子1-γ(anti-transcriptional intermediary 1γ antibody,T1F1-γ)蛋白。近期研究显示抗T1FI-γ抗体存在时,皮肌炎患者合并肿瘤的风险显著升高[4]。ADM发病女多于男,临床表现常见双上眼睑或眶周水肿性紫红色斑、甲周红斑及皮肤异色症,不伴有肌无力或肌痛,可伴有恶性肿瘤和肺部疾病,本文患者皮损表现与报道一致。
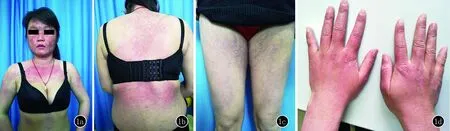
图11a:面部以眶周、两侧面颊为主的水肿性紫红斑,颈部V型区、双上肢近端皮肤散在红色斑片;1b:后背皮肤泛发红斑;1c:双股部散在暗红斑;1d:双手泛发Gottron丘疹

图22a:表皮轻度角化过度,基底层增厚,真皮浅层水肿,血管周少许单一核细胞浸润(HE,×200);2b:基底膜带IgG阳性带状沉积(免疫荧光,×200)
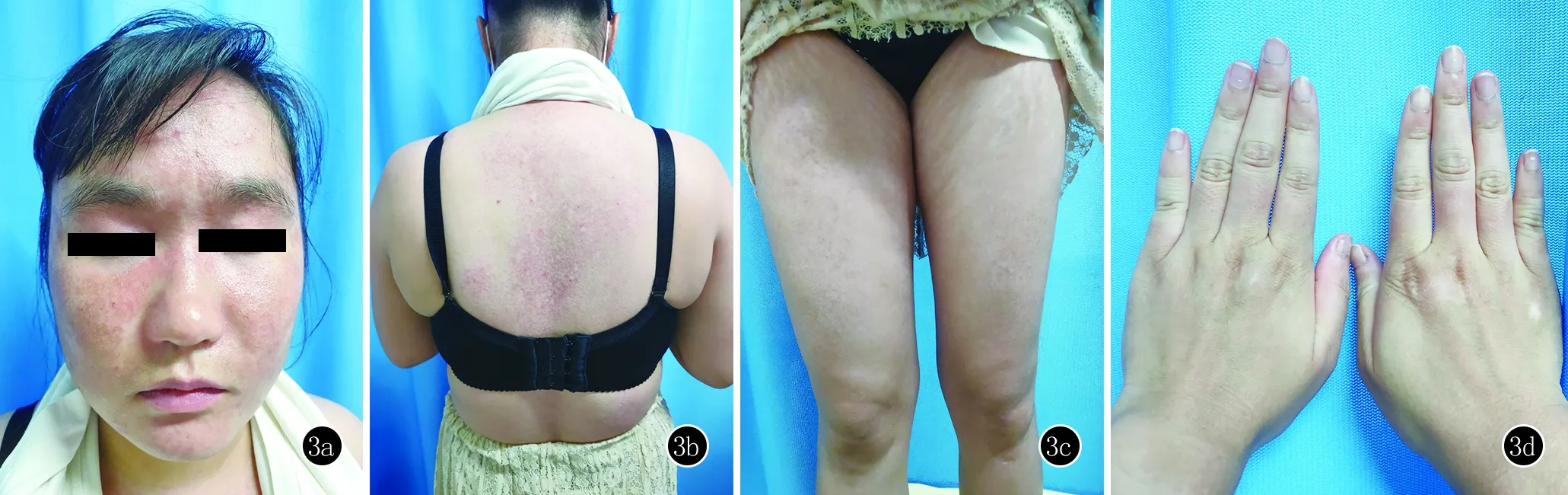
图33a:面部皮损基本消退;3b:后背皮肤红斑基本消退,留有色素沉着斑,中间伴有色素减退斑;3c:双股部皮肤异色症减轻,暗红斑减少;3d:双手Gottron丘疹大部分消退
Patel等[5]应用EULAR/ACR标准(Gottron丘疹、Gottron症、眼睑或眶周紫红色水肿性斑片并伴有眶周水肿)评价211例ADM患者,发现该标准适用于IIM诊断并提出同时加入活检组织病理评估ADM,提高ADM诊断率。目前临床上最常用Sontheimer[6]修正后的ADM诊断标准:(1)具有特征性皮肌炎表现,持续时间超过6个月;(2)无肌无力及肌酶谱的异常改变;(3)肌电图、肌肉活检及磁共振等肌肉检查均正常,同时除外发病6个月内经过连续2个月以上的免疫抑制剂治疗或使用羟基脲、他汀类降脂药等致皮肌炎样皮肤损害的药物。ADM组织病理学改变见表皮萎缩,灶性角化不全,常有基底细胞空泡变性,表皮基膜增厚;真皮网状层内黏蛋白沉积伴真皮水肿,毛细血管扩张;真皮中上部血管周围淋巴细胞浸润,常见嗜色素细胞。抗黑素瘤分化相关基因5(melanoma differentiation associated gene-5,MDA-5)与快速进展性间质性肺炎相关,通过检测抗 MDA5 抗体不仅可以判断ADM、CADM是否合并肺间质病变,其还可作为伴发急进型肺间质病变的血清学标记。但皮肌炎特异性抗体如Jo-1及抗Mi-2抗体多阴性,在ADM中多不具有特异性。肺部功能检查包括CT、肺功能检查、肺活检等。结合临床表现、肌酶相关指标、自身抗体及组织病理学、相关肺部功能检查可确诊该病[7]。本文患者符合上述诊断标准,诊断为无肌病性皮肌炎明确。
ADM治疗一般包括三线治疗:一线治疗方案为糖皮质激素和或免疫抑制剂,环孢素及环磷酰胺对ILD-ADM患者有一定治疗价值,特别是在呼吸衰竭前使用;二线为静脉注射人免疫球蛋白;三线为英夫利昔单抗。曹华等[10]对16例ADM患者长期随访显示2例患者进展为典型的皮肌炎,1例转归为慢性皮肤型红斑狼疮,2例并发内脏肿瘤,11例出现间质性肺炎的影像学改变。Suda等[11]通过分析14例ILD-ADM患者得出结论:急性、亚急性ILD-ADM患者病情进展迅速,治疗效果差,死亡率高;而慢性ILD-ADM治疗效果相对较好,预后良好。本文患者起初给予雷公藤及甲氨蝶呤治疗,未能控制病情,后给予泼尼松联合甲氨蝶呤治疗后,病情控制,皮损明显消退。治疗后近半年患者按时复诊,病情控制良好。
无肌病性皮肌炎临床少见,本文患者青年女性,病史较长,眶周紫红色水肿性斑片、皮肤异色症及双手Gottron丘疹等临床表现典型,结合组织学病理且肌酶基本正常,无近端肌无力症状,诊断明确。患者多次复诊,应用糖皮质激素联合免疫抑制剂,病情控制良好。胸片无异常,且MDA5抗体及肿瘤标记物均阴性,提示患者当前并发ILD概率较低,但患者抗T1FI-γ抗体阳性,预后仍需要继续随访观察。
猜你喜欢
现代临床医学(2022年2期)2022-04-19
南京中医药大学学报(2021年2期)2021-12-24
医学前沿(2021年6期)2021-09-10
现代临床医学(2021年4期)2021-07-31
——脂溢性皮炎样皮肌炎
中国临床医学(2021年1期)2021-03-03
中国生殖健康(2020年2期)2021-01-18
世界最新医学信息文摘(2020年18期)2020-12-24
中国医学创新(2020年7期)2020-05-06
祝您健康·养生堂(2019年1期)2019-09-10
中国中医药信息杂志(2018年10期)2018-12-20
