
一测多评法同时测定宽中顺气丸中8 种成分
2020-04-29 06:00李华生王文渊
中成药 2020年4期
骆 航,李华生,王文渊
(永州职业技术学院医学院,湖南 永州 425100)
宽中顺气丸由香附(醋炙)、陈皮、莪术(醋炙)、三棱(麸炒)、木香、五灵脂(醋炙)、猪牙皂、黄芩、牵牛子、大黄、滑石11 味药材组成,临床上主要用于气血郁滞、停食停水引起的胸膈痞满、膨闷胀饱、不欲饮食、脘腹胀痛、大便秘结等病症的治疗[1]。方中香附(醋炙)疏肝解郁,理气宽中,为君药;辅以木香、陈皮行气燥湿,健脾消食;五灵脂(醋炙)活血化瘀;莪术(醋炙)、三棱(麸炒)破血行气,消积止痛;猪牙皂开窍散结;黄芩、大黄清热燥湿,泻下攻积;牵牛子、滑石利尿通淋,泻水通便,引药下行,诸药合用,共奏顺气宽胸、消积化滞之功效[2]。
中药及其制剂组成复杂,通过多成分、多靶点协同作用而达到治疗效果,单一成分检测难以全面评价其整体质量,多指标成分评价模式已逐步应用于相关质量控制中,但传统方法存在消耗大、检验成本高、部分对照品质量不稳定并难以获得等诸多不足。一测多评法利用中药有效成分间存在的内在函数关系,建立内标与其他成分之间的相对校正因子,仅使用1 种质量稳定、易于获得、价格低廉的对照品,实现对中药及其制剂中多指标成分含有量的同时测定,正成为质量评价新模式。本实验选择宽中顺气丸中君药香附(醋炙)所含香附烯酮、α-香附酮,臣药陈皮所含橙皮苷、川陈皮素、桔皮素,佐药莪术(醋炙)所含莪术二酮、莪术醇、吉马酮作为指标成分,建立一测多评法同时测定这8 种成分含有量,以期为完善和提高该制剂质量标准提供数据支持。
1 材料
Agilent 1100 型、Waters 2690 型高效液相色谱仪(美国Waters 公司);BP211DAG 型电子天平(德国Sartorius 公司)。吉马酮(批号111665-201605,纯度99.8%)、莪术二酮(批号111800-201302,纯度99.8%)、橙皮苷(批号110721-201818,纯度96.2%)、莪术醇(批号100185-201007,纯度99.9%)、α-香附酮(批号110748-201815,纯度99.7%)对照品均购自中国食品药品检定研究院;桔皮素对照品(批号PRF8063048,纯度98.7%)购自成都普思生物科技股份有限公司;川陈皮素(批号18030541,纯度99.9%)、香附烯酮(批号18051522,纯度95.5%)对照品均购自上海同田生物技术股份有限公司;木香、陈皮、香附(醋炙)、三棱(麸炒)、莪术(醋炙)、五灵脂(醋炙)、猪牙皂、黄芩、牵牛子、大黄、滑石均购自永州市永靛中药饮片有限公司,经永州职业技术学院医学院王文渊教授鉴定为正品,按各药材质量标准检验均符合规定。宽中顺气丸(每100 粒重6 g,批号18080001、18080016、19010024)来源于北京同仁堂股份有限公司同仁堂制药厂。乙腈为色谱纯;其余试剂均为分析纯。
2 方法与结果
2.1 溶液制备
2.1.1 对照品溶液 精密称取橙皮苷、川陈皮素、桔皮素、莪术二酮、莪术醇、吉马酮、香附烯酮、α-香附酮对照品适量,甲醇制成质量浓度分别为0.914、0.198、0.172、0.318、0.206、0.188、1.352、0.474 mg/mL 的贮备液,各精密吸取2.5 mL置于50 mL 量瓶中,甲醇稀释至刻度,摇匀,即得(橙皮苷45.7 μg/mL、川陈皮素9.9 μg/mL、桔皮素8.6 μg/mL、莪术二酮15.9 μg/mL、莪术醇10.3 μg/mL、吉马酮9.4 μg/mL、香附烯酮67.6 μg/mL、α-香附酮23.7 μg/mL)。
2.1.2 供试品溶液 取丸剂适量,研细,精密称取2.0 g,置于具塞锥形瓶中,精密加入25 mL 甲醇,称定质量,加热回流提取60 min,放冷,甲醇补足减失的质量,摇匀,滤过,即得。
2.1.3 阴性样品溶液 按处方比例和工艺,分别制备缺陈皮、缺莪术、缺香附的阴性样品,按“2.1.3”项下方法制备,即得。
2.2 色谱条件及系统适用性试验 Agilent TC-C18色谱柱(250 mm×4.6 mm,5 μm);流动相乙腈(A)-0.1%冰醋酸(B),梯度洗脱,程序见表1;体积流量1.0 mL/min;检测波长326 nm(0~27.0 min,橙皮苷、川陈皮素、桔皮素)[3-5]、214 nm(27.0~45.0 min,莪术二酮、莪术醇、吉马酮)[6-9]、242 nm(45.0~70.0 min,香附烯酮、α-香附酮)[10-11];柱温25 ℃;进样量10 μL。取对照品、供试品、阴性样品溶液适量,在上述色谱条件下进样测定,结果见图1,可知各成分与相邻色谱峰均能达到有效分离,分离度均大于1.5,理论塔板数按照各成分色谱峰计均不低于5 000,阴性无干扰。
2.3 方法学考察
2.3.1 线性关系考察 精密量取“2.1.1”项下对照品贮备液适量,甲醇制成25 倍质量浓度差的6 个对照品溶液,在“2.2”项色谱条件下进样测定。以峰面积为纵坐标(Y),溶液质量浓度为横坐标(X)进行回归,结果见表2,可知各成分在各自范围内线性关系良好。
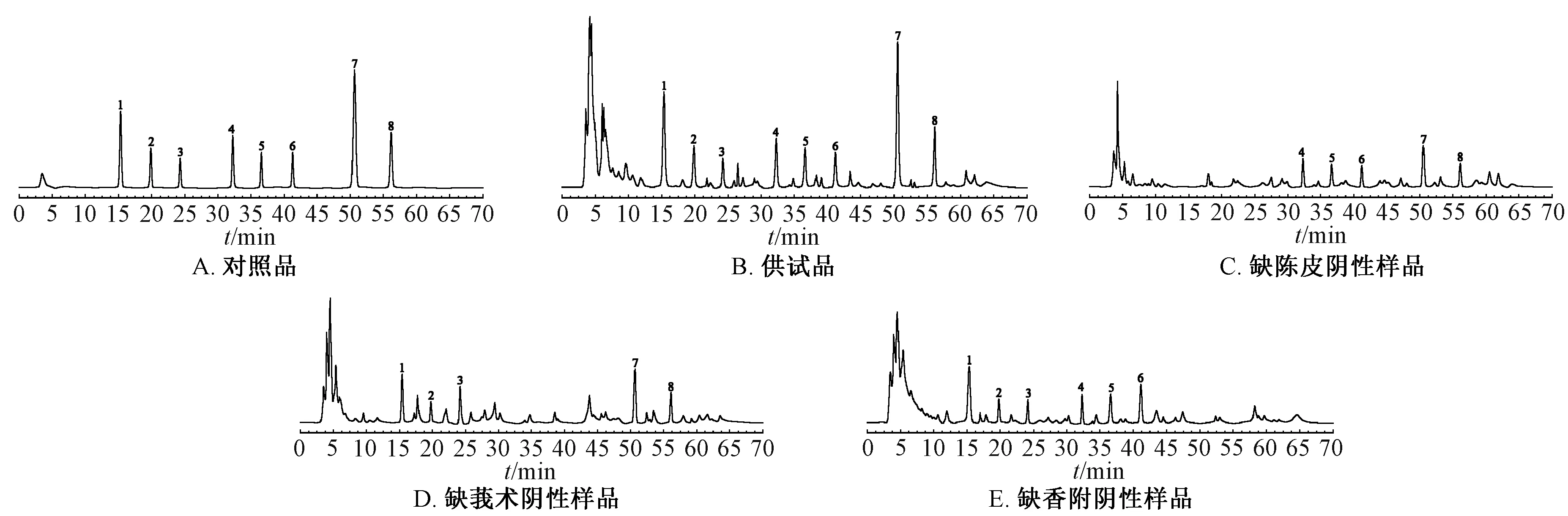
图1 各成分HPLC 色谱图Fig.1 HPLC chromatograms of various constituents
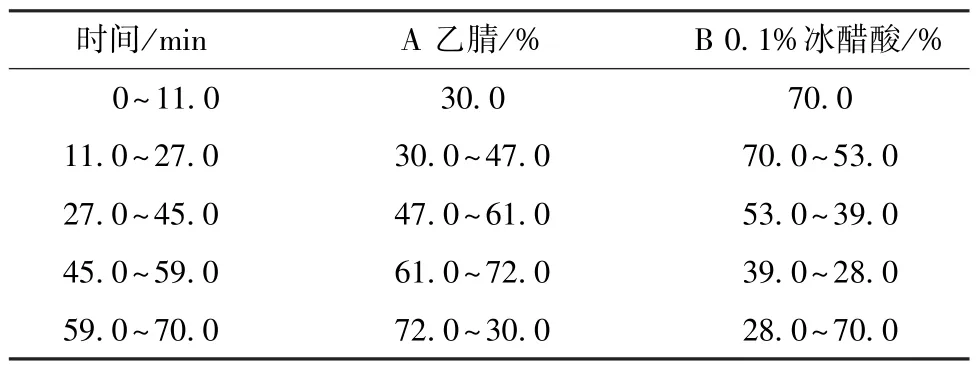
表1 梯度洗脱程序Tab.1 Gradient elution programs

表2 各成分线性关系Tab.2 Linear relationships of various constituents
2.3.2 精密度试验 精密吸取“2.1.1”项下对照品溶液适量,在“2.2”项色谱条件下进样测定,测得橙皮苷、川陈皮素、桔皮素、莪术二酮、莪术醇、吉马酮、香附烯酮、α-香附酮峰面积RSD分别为0.66%、0.94%、1.15%、1.02%、1.05%、1.23%、0.58%、0.72%,表明仪器精密度良好。
2.3.3 重复性试验 取同一批丸剂,按“2.1.2”项下方法平行制备6 份供试品溶液,在“2.2”项色谱条件下进样测定,测得橙皮苷、川陈皮素、桔皮素、莪术二酮、莪术醇、吉马酮、香附烯酮、α-香附酮含有量RSD 分别为1.01%、0.67%、1.35%、1.29%、1.46%、1.78%、0.92%、1.17%,表明该方法重复性良好。
2.3.4 稳定性试验 取同一份供试品溶液,于0、2、4、6、8、12 h 在“2.2”项色谱条件下进样测定,测得橙皮苷、川陈皮素、桔皮素、莪术二酮、莪术醇、吉马酮、香附烯酮、α-香附酮峰面积RSD分别为0.59%、1.02%、1.18%、1.05%、1.12%、1.27%、0.53%、0.69%,表明溶液在12 h 内稳定性良好。
2.3.5 加样回收率试验 取同一批各成分含有量已知的丸剂适量,研细,精密称取9 份,每份1.0 g,随机分成3 组,每组3 份,置于具塞锥形瓶中,精密加入对照品溶液(橙皮苷、川陈皮素、桔皮素、莪术二酮、莪术醇、吉马酮、香附烯酮、α-香附酮质量浓度分别为 0.678、0.144、0.098、0.182、0.136、0.112、0.938、0.322 mg/mL )0.5、1.0、1.5 mL,使目标成分加入量分别约为样品原有量的50%、100%、150%,按“2.1.2”项下方法制备供试品溶液,在“2.2”项色谱条件下进样测定,计算回收率。结果,橙皮苷、川陈皮素、桔皮素、莪术二酮、莪术醇、吉马酮、香附烯酮、α-香附酮平均加样回收率(RSD)分别为98.32%(1.32%)、97.87%(1.63%)、96.93%(1.20%)、98.87%(0.99%)、98.52%(0.81%)、99.74%(0.97%)、100.00%(1.03%)、99.03%(1.11%)。
2.4 相对校正因子测定 取“2.3.1”项下对照品溶液适量,在“2.2”项色谱条件下进样测定,以吉马酮为内标,测定其他7 种成分相对校正因子fk/m,公式为fk/m=fk/fm=(Wk×Am)/(Wm×Ak)(Wk为内标含有量,Ak为内标峰面积,Wm为待测成分含有量,Am为待测成分峰面积),结果见表3。
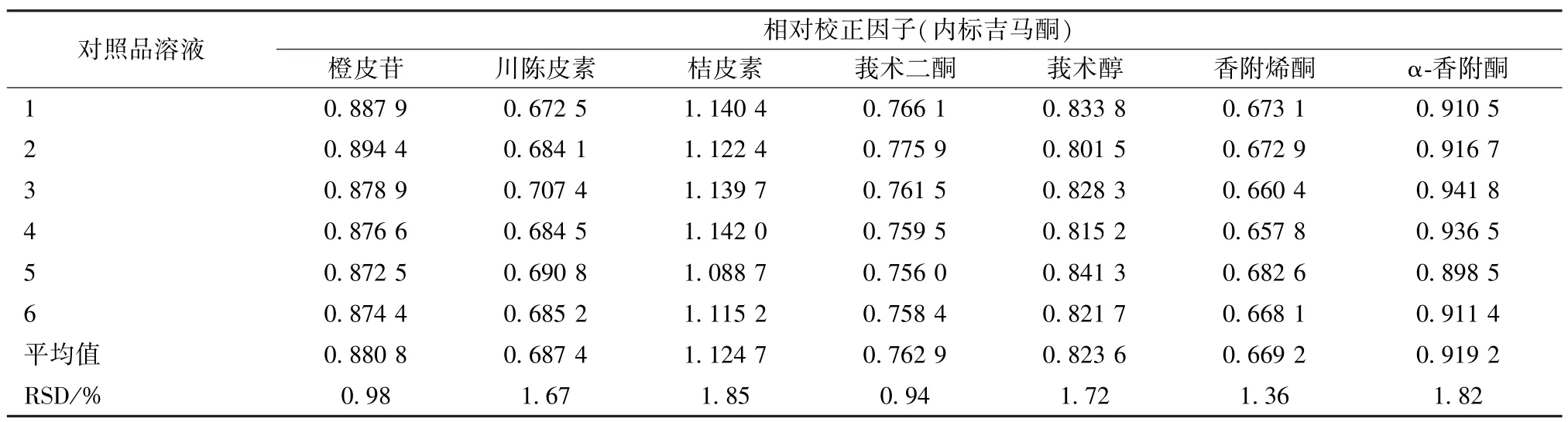
表3 各成分相对校正因子Tab.3 Relative correction factors of various constituents
2.5 耐用性考察
2.5.1 不同仪器、色谱柱对相对校正因子的影响 本实验考察了Waters 2690、Agilent 1100 色谱仪,以及Agilent TC-C18、Diamonsil C18、Phenomenex C18色谱柱(250 mm×4.6 mm,5 μm)对相对校正因子的影响,结果见表4,可知均无明显影响(RSD<2.0%)。
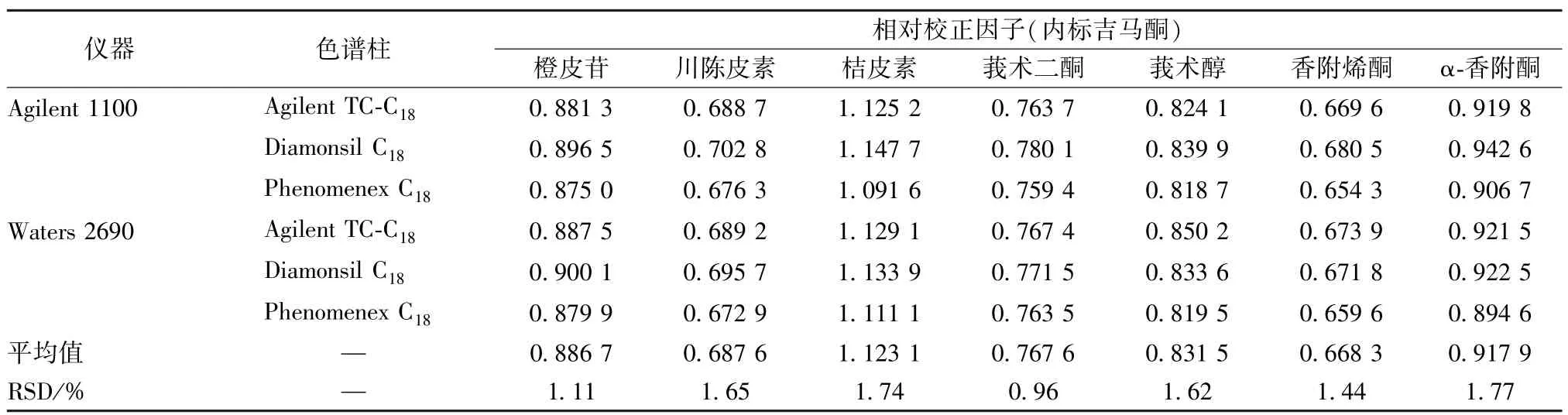
表4 不同仪器、色谱柱对相对校正因子的影响Tab.4 Effects of different instruments and columns on relative correction factors
2.5.2 不同体积流量对相对校正因子的影响 本实验考察了不同体积流量(0.8、0.9、1.0、1.1、1.2 mL/min)对相对校正因子的影响,结果见表5,可知均无明显影响(RSD<2.0%)。
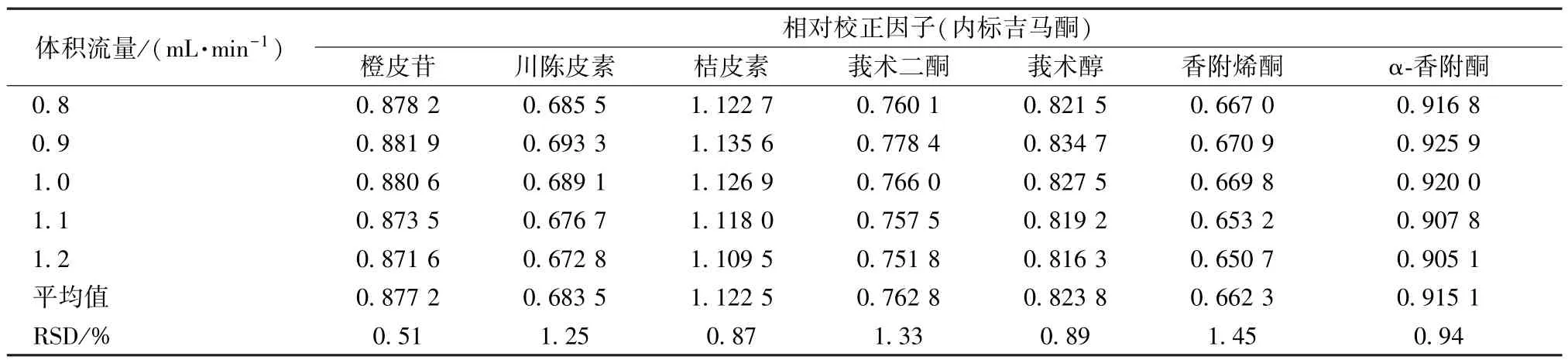
表5 不同体积流量对相对校正因子的影响Tab.5 Effects of different volumetric flow rates on relative correction factors
2.5.3 不同柱温对相对校正因子的影响 本实验考察了不同柱温(20、22、25、27、30 ℃)对相对校正因子的影响,结果见表6,可知均无明显影响(RSD<2.0%)。
2.6 色谱峰定位 本实验以吉马酮为内标,采用相对保留时间法(待测成分与内标保留时间之比)对其他成分色谱峰进行定位,考察了“2.5.1”项下仪器、色谱柱对相对保留时间的影响,结果见表7,可知均无明显影响(RSD<2.0%)。
2.7 样品含有量测定 取3 批丸剂,按“2.1.3”项下方法平行制备3 份供试品溶液,在“2.2”项色谱条件下进样测定,分别采用外标法和一测多评法计算含有量,结果见表8,可知2 种方法所得结 果接近[相对平均偏差(RAD)<2.0%]。
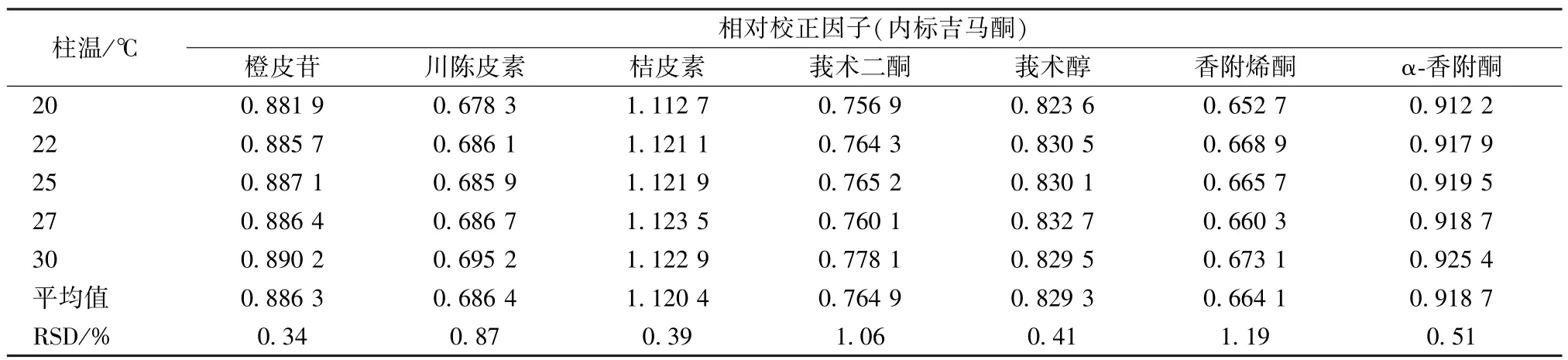
表6 不同柱温对相对校正因子的影响Tab.6 Effects of different column temperatures on relative correction factors
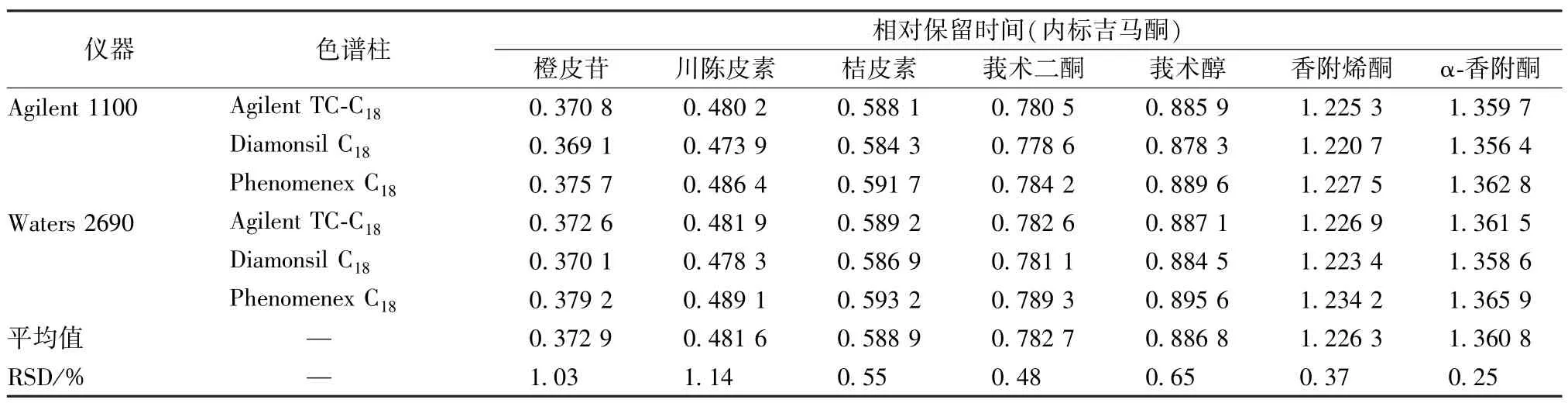
表7 不同仪器、色谱柱对相对保留时间的影响Tab.7 Effects of different instruments and columns on relative retention time
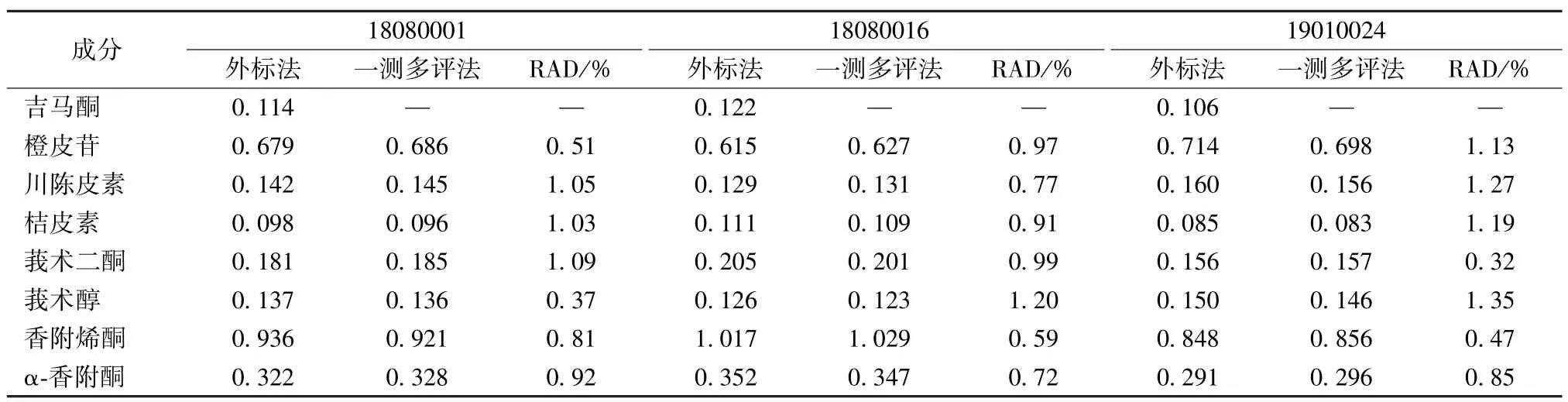
表8 各成分含有量测定结果(mg/g)Tab.8 Results of content determination of various constituents(mg/g)
3 讨论
3.1 色谱峰定位方法确定 本实验曾采用保留时间差法对宽中顺气丸中各成分进行定位,但发现不同仪器、色谱柱下其保留时间差差异较大,难以对色谱峰进行准确定位;相对保留时间值法所得结果稳定,不同条件下其RSD 均小于2.0%,故最终确定采用该方法进行色谱峰。
3.2 流动相确定 本实验考察了乙腈-水[6]、乙腈-0.1% 甲酸、乙腈-0.1% 磷酸[8]、乙腈-0.1% 冰醋酸[3-5,12]、甲醇-水[7,10-11,13-14]流动相体系,考察各成分峰形、分离度、色谱峰基线平稳情况,发现乙腈-0.1%冰醋酸洗脱时综合效果最佳,再对其不断进行改进优化,最终确定其作为流动相,此时各成分色谱图基线平稳,峰形对称,分离度符合规定。
4 结论
本实验首次建立一测多评法同时测定宽中顺气丸中橙皮苷、川陈皮素、桔皮素、莪术二酮、莪术醇、吉马酮、香附烯酮、α-香附酮的含有量,再通过方法学考察、相对校正因子耐用性试验、色谱峰定位、与外标法结果比较来验证其可行性。结果表明,该方法操作简便,结果准确,重复性好,可用于宽中顺气丸质量控制,为全面评价该制剂质量提供参考依据。
猜你喜欢
中国老年学杂志(2022年22期)2022-11-21
中国饲料(2022年5期)2022-04-26
食品安全导刊·中旬刊(2022年3期)2022-04-15
化学与生物工程(2020年6期)2020-06-30
中国海洋大学学报(自然科学版)(2019年10期)2019-10-14
中国兽医杂志(2017年10期)2017-12-01
中国医药指南(2017年3期)2017-11-13
中国药房(2017年28期)2017-10-13
妇女生活(2017年8期)2017-09-05
妇女生活(2016年9期)2016-09-08
