
端粒酶逆转录酶启动子热点突变的ARMS-LNA-qPCR检测方法建立
2020-04-29 14:30:28齐长海梁国威
中国实验诊断学 2020年4期
张 洁,李 方,齐长海,梁国威*
(1.北京大学航天临床医学院 检验科,北京100049;2.航天中心医院 病理科)
2013 年科学杂志首次报道了在黑色素瘤家系和患者中检测到端粒酶逆转录酶(TERT)启动子区域-124 bp(C/T,C228T)和-146 bp (C>T,C250T )高突变率(突变率>70%)[1],该热点突变在肝细胞肝癌(HCC)组织中的突变率高达60%[2],已成为肝癌及多种恶性肿瘤的诊断、治疗监测和预后评估的重要分子监测靶点[3,4]。
目前,检测TERT该热点突变的方法只有二代测序和液滴数字PCR(ddPCR)技术平台(突变检测下限分别为0.1%左右和0.1%-0.05%左右)2种方法[5,6],但皆存在不适用于临床常规开展等问题。我们以普通PCR扩增含有TERT热点突变位点的纯化产物作为检测标本,采用等位基因扩增阻滞原理(ARMS)的引物,并结合锁核酸(LNA)抑制探针,建立了实时荧光定量PCR检测TERT启动子热点突变率方法(ARMS-LNA-qPCR),并在30例表观健康人中确定检测下限。
1 对象与方法
1.1 研究对象
表观健康者:收集我院2019年3月-2019年5月健康体检确认的表观健康志愿者30例,纳入标准为:无乙肝、丙肝、药物肝、酒精肝病史或肝脏蜘蛛痣,实验室检查肝、肾功能正常,根据肿瘤标志物和影像学检查除外恶性肿瘤。其中男性16例,女性14例,年龄范围21-35岁,平均年龄29.1±3.8岁。本研究通过航天中心医院医学伦理委员会审查(项目批号:20190830-YN-01)。
1.2 表观健康人外周血基因组DNA
EDTA抗凝外周全血200 μl用于基因组DNA提取,采用天根生化科技(北京)有限公司的DNA提取试剂盒(目录号:DP304-02),按试剂盒说明操作。提取后的组织DNA立即-80℃冻存备用。
1.3 ARMS-LNA-qPCR方法建立
采用2步法建立TERT启动子区-124bp(C/T)和-146bp(C/T)突变率检测方法。第一步富集模板,普通PCR扩增产物纯化后作为待检标本;第二步突变率检测,以ARMS-LNA-qPCR中ΔCt值(ΔCt值=内参Ct值-突变率标准品Ct值)与突变率标准品建立的标准曲线计算获得。所建方法的检测原理、引物、探针及循环条件见国家知识产权局专利申请号201910991396.3。
1.4 直接测序
为了验证ARMS-LNA-qPCR方法的准确性,30例表观健康人全部采用直接测序法进行了测序(诺赛基因技术有限公司,北京,中国)。
1.5 统计学处理
2 结果
2.1 ARMS-LNA-qPCR定量检测TERT突变率标准曲线
采用所建ARMS-LNA-qPCR方法,分别检测突变率标准品为100%、10%、1%、0.1%、0.05%和0%(100%W)的-124bp(C/T)和-146bp(C/T)突变率,其突变率扩增曲线及△Ct与Lg突变率回归曲线见图1。
对于-124位点,ARMS-LNA-qPCR突变率扩增曲线图(重复2次)显示检测突变率的下限达到0.05%(Y=-3.801X+33.02,R2=0.999,P<0.001),其ΔCt(0.05%M-100%W)=-2.11,这一差值区间可以显著区分0.05%突变和100%野生样本。为了监控富集PCR产物质量并校准紫外分光光度计测定浓度所引起误差,我们采用1011拷贝数/ml突变率标准品重复测定5次,建立了△Ct 值(内参qPCR的Ct值减去ARMS-LNA-qPCR的Ct值)与-124位点突变率的内参突变率标准曲线,结果显示Lg-124突变率与△Ct 值呈线性关系(P<0.001,R2=0.999),其突变率回归方程为-124突变率= 10(0.262×△Ct+4.325),见图1(A-1和A-2)。
对于-146位点,ARMS-LNA-qPCR突变率扩增曲线图(重复2次)显示检测突变率的下限达到0.05%(Y=-3.834X+32.097,R2=0.999,P<0.001),其ΔCt(0.05%M-100%W)=-1.92,这一差值区间可以显著区分0.05%突变和100%野生样本。采用1011拷贝数/ml突变率标准品重复测定5次,结果显示Lg-146突变率与△Ct 值呈线性关系(P<0.001,R2=0.999),其突变率回归方程为-146突变率= 10(0.261△Ct+3.895),见图1(B-1和B-2)。
2.2 ARMS-LNA-qPCR方法检测下限的判定
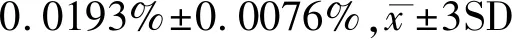
3 讨论
本研究在ARMS-qPCR的基础上,采用LNA抑制探针选择性抑制ARMS-qPCR中突变引物对野生等位基因的非特异性扩增,成功建立了高敏感定量检测TERT启动子-124bp和-146bp位点突变率的ARMS-LNA-qPCR方法。该方法定量检测-124bp和-146bp位点突变率的下限是0.07%和0.05%。
ARMS-LNA-qPCR方法成立的关键是LNA抑制探针对野生和突变等位基因的结合具有极强的选择性。研究显示,寡核苷酸中1个碱基进行LNA修饰可增加其Tm值3-8℃,如果错配发生在LNA碱基上,其与DNA亲和力则明显下降,利用这一特性可明显增加LNA修饰探针对等位基因的区分能力[7]。我们在ARMS-qPCR中逐渐增加LNA抑制探针的浓度,通过比较100%和0%TERT-124和-146突变标准品Ct值的变化,最终选择0.25 μM(-124位点)和0.2 μM(-146位点)终浓度的LNA抑制探针作为抑制野生等位基因扩增的合适水平。需要注意的是,由于各实验室条件不尽相同,建议不同实验室注意LNA抑制探针浓度的优化。
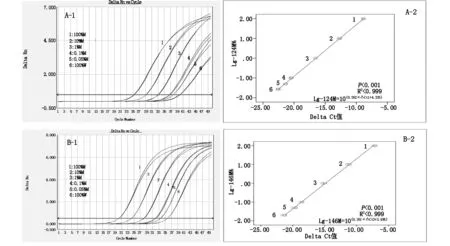
图1 ARMS-LNA-qPCR方法测定TERT-124bp(C/T) 和-146bp(C/T)位点突变率标准品的扩增曲线及Lg突变率-△Ct值回归曲线图
A-1:1011拷贝数/ml突变率标准品扩增曲线图,ΔCt(0.05%M-100%W)=-2.11 ;A-2:Lg-124突变率与ΔCt值(124内参qPCR的Ct值减去-124 ARMS-LNA-qPCR的Ct值)的突变率回归曲线;B-1:1011拷贝数/ml突变率标准品扩增曲线图,ΔCt(0.05%M-100%W)=-1.92; B-2:Lg-146突变率与ΔCt值(124内参qPCR的Ct值减去-146 ARMS-LNA-qPCR的Ct值)的突变率回归曲线。
我们建立的ARMS-LNA-qPCR方法,采用PCR纯化产物在qPCR技术平台上进行检测,其TERT-124bp和-146bp位点突变率的检测下限为0.07%和0.05%,与二代测序和ddPCR检测下限基本相同。由于qPCR技术平台是目前临床常规分子检测技术平台,且所建方法采用PCR扩增纯化标本进行检测,因此ARMS-LNA-qPCR方法具有临床广泛开展的适用性和多种标本来源的普适性。
综上所述,本研究建立的ARMS-LNA-qPCR方法灵敏度较高,其敏感性和重复性皆可满足临床需要。由于采用qPCR技术平台和PCR扩增纯化标本进行检测,该方法具有广泛的临床应用价值。
猜你喜欢
遗传(2021年10期)2021-11-01 10:30:08
河北医学(2021年10期)2021-10-27 00:37:14
矿山安全信息(2021年21期)2021-07-04 06:33:32
矿山安全信息(2020年37期)2020-12-26 07:25:58
中国医学创新(2020年11期)2020-06-08 10:38:37
中华肺部疾病杂志(电子版)(2020年2期)2020-05-07 00:34:24
矿山安全信息(2020年2期)2020-03-05 05:13:56
矿山安全信息(2020年3期)2020-03-04 10:18:08
中国临床医学影像杂志(2019年6期)2019-08-27 02:59:50
发明与创新(2015年25期)2015-02-27 10:39:16
