
Sequencing analysis of matrix metalloproteinase 7-induced genetic changes in Schwann cells
2020-04-29 02:12PanJianLuGangWangXiaoDongCaiPingZhangHongKuiWang
中国神经再生研究(英文版) 2020年11期
Pan-Jian Lu, Gang Wang, Xiao-Dong Cai, Ping Zhang, Hong-Kui Wang
Key Laboratory of Neuroregeneration of Jiangsu and Ministry of Education, Co-innovation Center of Neuroregeneration, Nantong University,Nantong, Jiangsu Province, China
Abstract Previous research revealed the positive activity of matrix metalloproteinase 7 (MMP7) on migration and myelin regeneration of Schwa nn cells (SCs). However, understanding of the molecular changes and biological activities induced by increased amounts of MMP7 in SCs remains limited. To better understand the underlying molecular events, primary SCs were isolated from the sciatic nerve stump of newborn rats and cultured with 10 nM human MMP7 for 24 hours. The results of genetic testing were analyzed at a relatively relaxed threshold value (fold change ≥ 1.5 and P-value < 0.05). Upon MMP7 exposure, 149 genes were found to be upregulated in SCs, whereas 133 genes were downregulated. Gene Ontology analysis suggested that many differentially expressed molecules were related to cellular processes, single-organism processes, and metabolic processes. Kyoto Enrichment of Genes and Genomes pathway analysis further indicated the critical involvement of cell signaling and metabolism in MMP7-induced molecular regulation of SCs. Results of Ingenuity Pathway Analysis (IPA)also revealed that MMP7 regulates biological processes, molecular functions, cellular components, diseases and functions, biosynthesis,material metabolism, cell movement, and axon guidance. The outcomes of further analysis will deepen our comprehension of MMP7-induced biological changes in SCs. This study was approved by the Laboratory Animal Ethics Committee of Nantong University, China(approval No. 20190225-004) on February 27, 2019.
Key Words: bioinformatic analysis; ingenuity pathway analysis; matrix metalloproteinase 7; peripheral nerve regeneration; RNA sequencing;Schwann cells; sciatic nerve injury
Introduction
Matrix metalloproteinases (MMPs), also called matrixins,are members of a family of calcium-dependent zinc-containing endopeptidases (Verma and Hansch, 2007). MMPs function extracellularly and are capable of degrading extracellular matrix components and cell surface proteins by affecting a large series of proteolytic events (Zhang et al., 2011;Mirzaie et al., 2018). The proteolytic capabilities of MMPs make them critical for tissue remodeling, repair, and regeneration, as well as wound healing, in both physiological and pathological circumstances (Visse and Nagase, 2003; Paiva and Granjeiro, 2014; Ma et al., 2020).
Differential expression or activation of MMPs, or both,has been associated with many pathological conditions in the nervous system, including Alzheimer’s disease, multiple sclerosis, ischemia/reperfusion, Parkinson’s disease, and central nervous system injury (Agrawal et al., 2008; Kim and Joh, 2012; Beroun et al., 2019). In addition, recent studies have shown that MMPs are also altered after peripheral nervous system injury (Qin et al., 2016; Yi et al., 2017; Remacle et al., 2018). MMP7, an MMP family member also called matrilysin, pump-1 protease, or uterine metalloproteinase,was dramatically upregulated in the two nerve ends of rats after sciatic nerve crush injury (Qin et al., 2016). In a previous study, we evaluated the biological activities of MMP7 in peripheral nerve regeneration by determining its effect on Schwann cells (SCs), the main glial cells in the peripheral nervous system. Functional analysis suggested that increased MMP7 encouraged SC migration and the formation of myelin sheaths. Moreover, mechanisms underlying the observed effects of MMP7 on SCs were preliminary examined by RNA sequencing (Wang et al., 2019).
RNA sequencing is a powerful high-throughput experimental technology for data mining and gene searching, as it provides a simple way to characterize the abundances of thousands of genes in one assay (Nagalakshmi et al., 2010;Stefaniuk and Ropka-Molik, 2016). In our previous study, we compared gene abundances in primary SCs cultured with or without exogenous MMP7 protein, and screened a total of 34 differentially expressed genes with a fold change ≥ 2 andP-value < 0.05 (Wang et al., 2019). This provided some clues about MMP7-induced genetic modifications in SCs; however, considering the small amount of identified differentially expressed genes, the information obtained was limited and some critical details may have been overlooked.
To achieve a more comprehensive understanding of MMP7-induced genetic changes, genes with relatively less robust differential expression might be included and investigated. Besides a fold change of 2, a fold change of 1.5 is also commonly used as a threshold value (Gong et al.,2016). Herein, we used the edgeR package to re-set a lower threshold value (fold change ≥ 1.5 andP-value < 0.05), with the aim of identifying affected genes with less significant alterations of expression. Bioinformatic tools including Gene Ontology (GO) functions, Kyoto Enrichment of Genes and Genomes (KEGG) pathway analysis, and Ingenuity Pathway Analysis (IPA) were applied to analyze the biological meanings of these genetic changes.
Materials and Methods
Ethics statement
A total of 30 neonatal female Sprague-Dawley rats (1-day old) were acquired from the Experimental Animal Center of Nantong University [License No. SCXK (Su) 2014-0001 and SYXK (Su) 2012-0031, No. 20190225-004] for sciatic nerve collection and SC isolation. Animal procedures were approved by the Laboratory Animal Ethics Committee of Nantong University, China (No. 20190225-004) on February 27,2019 and conducted in accordance with Institutional Animal Care Guidelines of Nantong University (Nantong, China).
Isolation of SCs
Primary SCs were obtained from the sciatic nerve stumps of newborn rats (Qian et al., 2018). Briefly, sciatic nerves were carefully dissected, collected, cut, and digested for isolation and further culture of primary SCs. Mouse anti-Thy1.1 antibody (1:1000; Sigma, St. Louis, MO, USA) and rabbit complement sera (10 mg/mL; Sigma) were added to remove contaminating fibroblasts. Rabbit anti-S100 antibody (1:200;Dako, Carpinteria, CA, USA) and secondary goat anti-rabbit antibody (1:100; Abcam, Cambridge, UK) were used to determine the purity of SCs by fluorescence microscopy (Axio-Imager M2, Zeiss, Jena, Germany; Additional Figure 1).
SCs were cultured in Dulbecco’s Modified Eagle Medium (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (Invitrogen), 1% penicillin and streptomycin(Invitrogen), 2 μM forskolin (Sigma), and 10 ng/mL human heregulin-β1 (Sigma). SCs were incubated in a humidified 5% CO2incubator at 37°C. Medium was refreshed every other day. Primary SCs were exposed to 10 nM recombinant human MMP7 purified protein (Merck Millipore, Billerica,MA, USA) for 24 hours prior to RNA isolation.
RNA sequencing
Total RNA was isolated from primary SCs using TRIzol(Invitrogen). RNA sequencing was conducted by Illumina HiSeqTM(Gene Denovo Biotechnology Co., Guangzhou,China) as previously described (Wang et al., 2019). Eukaryotic mRNA was enriched by magnetic beads with Oligo(dT),fragmented into short pieces, reverse transcribed into cDNA,and ligated to Illumina sequencing adapters. Raw reads containing adapters or low-quality bases were filtered. Clean reads were aligned to a reference genome to obtain gene annotations and abundances. Gene abundances were quantified by RNA-Seq by Expectation Maximization (RSEM) software v1.3.1 (University of Wisconsin-Madison, Madison, WI,USA) (Li and Dewey, 2011) and normalized as fragments per kilobase of transcript per million mapped reads (FPKM):FPKM = 106C/(NL/103), where C indicates the number of fragments mapped to the target gene, N indicates the total number of fragments mapped to reference genes, and L indicates the number based on target gene. RNA-seq data have been deposited in the National Center for Biotechnology Information Sequence Red Archive under the accession code SRP173072.
Bioinformatic analysis
The edgeR package (http://www.r-project.org/) was used to identify differentially expressed genes in the MMP7-treated group compared with the untreated control group. Genes with a fold change ≥ 1.5 andP-value < 0.05 were considered significant differentially expressed genes and are listed in
Additional Table 1.
Differential genes were analyzed with Database for Annotation, Visualization, and Integrated Discovery (DAVID;https://david.ncifcrf.gov/) bioinformatic resources and subjected to enrichment analysis for GO terms (http://www.geneontology.org) and KEGG pathways (http://www.genome.ad.jp/kegg/). The number of differentially expressed genes, including upregulated and downregulated genes, are displayed for enriched biological processes, molecular functions, cellular components, and signaling pathways. Furthermore, core analysis of differential molecules was conducted with IPA software (Ingenuity Systems, Redwood City, CA,USA). Mapped IDs were subjected to IPA for diseases and functions, networks, and canonical signaling pathways. Statistically significant diseases and functions, canonical pathways, networks, and the involved molecules involved were identified.
Results
Overview of differentially expressed genes in MMP7-induced SCs
To obtain a more comprehensive view of MMP7-induced genetic changes, a relatively relaxed threshold value (fold change ≥ 1.5 andP-value < 0.05) was set in the current study compared with our previous study (Wang et al., 2019).A larger number of molecules exhibited significant changes in expression at this threshold value (listed in Additional Table 1). A total of 149 genes were found to be upregulated in SCs in response to the addition of MMP7 protein.In addition, 133 genes were found to be downregulated in MMP7-treated SCs. Genes with significantly different expression in three biological replicates of MMP7-treated SCs(MMP7-1, MMP7-2, and MMP7-3) and negative control SCs (Con-1, Con-2, and Con-3) are displayed in the heatmap in Figure 1.
Identification of critical GO functions and KEGG pathways
The DAVID database was applied to annotate functions and signaling pathways of the 282 up-/down-regulated genes.Most differentially expressed molecules were categorized to the GO biological process terms “cellular process”, “single-organism process”, and “metabolic process”. With regard to GO molecular function terms, most differentially expressed genes were categorized to “binding and catalytic activity”. For GO cellular component terms, most differentially expressed genes were categorized to “cell”, “cell part”, and“organelle” (Figure 2). Enriched KEGG pathways included“organismal systems”, “environmental information process”,“metabolism”, “human diseases”, and “genetic information processing” (Figure 3). Consistent with the critical involvement of cellular and metabolic processes in GO biological process terms, KEGG outcomes showed that many differentially expressed genes were involved in signal transduction,signaling molecules and interactions, and metabolism.
Identification of critical IPA diseases and biological functions and networks
IPA software was also applied to reveal biological changes after MMP7 treatment. Gene IDs of differentially expressed genes were recognized by IPA software for subsequent core analysis. A total of 211 differentially expressed genes were mapped to the IPA database and analyzed by Ingenuity Pathways Knowledge Base. IPA diseases and functions were ranked byP-value and top 20 of these are listed in Figure 4. These top-enriched IPA diseases and biological functions could be mainly grouped to cellular behavior (cell death and survival, cellular compromise, carbohydrate metabolism,molecular transport, cellular movement, immune cell trafficking, inflammatory response), organ maintenance and development (endocrine system development and function,organ morphology, organismal development, hematological system development and function, connective tissue development and function), and dysfunction and disease(endocrine system disorders, organismal injury and abnormalities, connective tissue disorders, development disorder,hematological disease, infectious diseases, and inflammatory disease).
IPA networks were also built to clarify the interactions of differentially expressed genes as well as their biological functions. A total of nine IPA networks were constructed (Table 1). The top three IPA networks obtained a high score ≥ 50,indicating that the possibility of genes in these IPA networks not being connected was equal to or less than 10-50 (Lu et al., 2018). These top IPA networks were principally associated with cellular behavior, including cell death and survival,cellular compromise, cellular growth and proliferation, and cell-to-cell signaling and interaction.
Identification of critical IPA canonical signaling pathways
A total of 18 IPA canonical signaling pathways were identified for differentially expressed genes (Figure 5). The most enriched IPA molecular regulatory pathways were “cellular stress and injury (type II diabetes mellitus signaling and ultraviolet C-induced mitogen-activated protein kinase signaling)”, “cellular immune response (granzyme B signaling, tumoricidal function of hepatic natural killer cells, and cytotoxic T lymphocyte-mediated apoptosis of target cells)”,“organismal growth and development (axonal guidance signaling, alanine biosynthesis III, planar cell polarity pathway, human embryonic stem cell pluripotency, coenzyme A biosynthesis, proline biosynthesis, molybdenum cofactor biosynthesis, and factors promoting cardiogenesis in vertebrates)”, “infectious diseases (guanine and guanosine salvage I)”, “disease-specific pathways and pathogen-influenced signaling (basal cell carcinoma signaling and mechanisms of vi-ral exit from host cells)”, and “cell cycle regulation (ceramide signaling)”.

Table 1 Enriched Ingenuity Pathway Analysis networks
Enriched IPA canonical signaling pathways were overlapped to observe their interactions (Figure 6). “Axonal guidance signaling” was located in the center of the overlapped pathway network and strongly associated with other signaling pathways, such as “human embryonic stem cell pluripotency”, “factors promoting cardiogenesis in vertebrates”, “ceramide signaling”, and “ultraviolet C-induced mitogen-activated protein kinase signaling”.
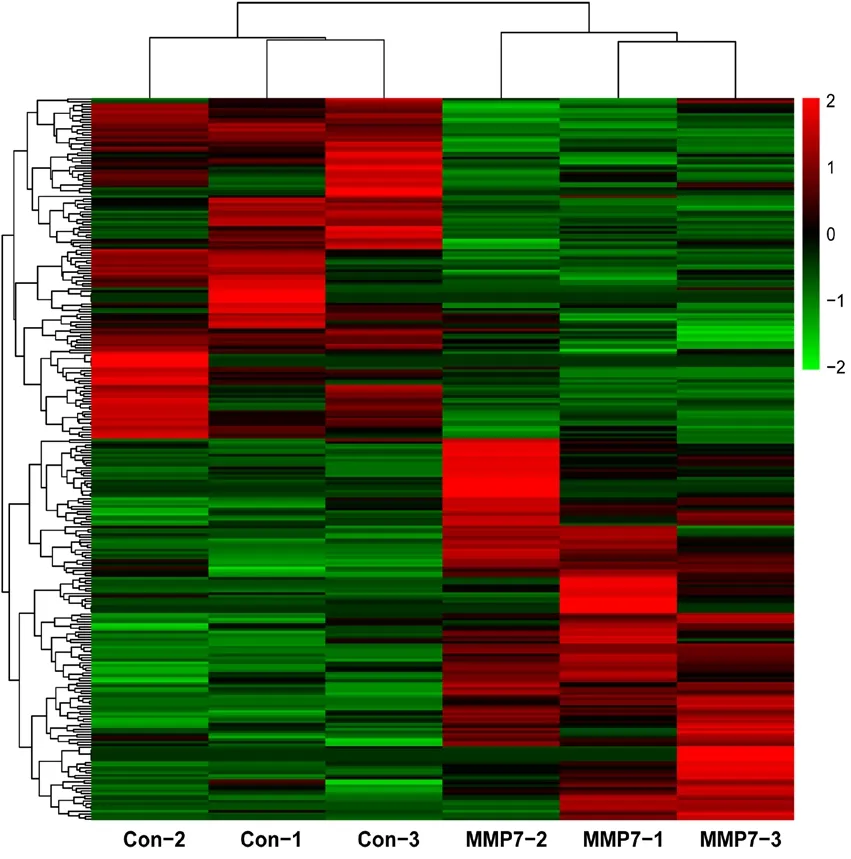
Figure 1 Heatmap of differentially expressed genes in MMP7-induced Schwann cells.

Figure 2 Enriched Gene Ontology (GO) biological process, molecular function, and cellular component terms for differentially expressed genes in matrix metalloproteinase 7-induced Schwann cells.
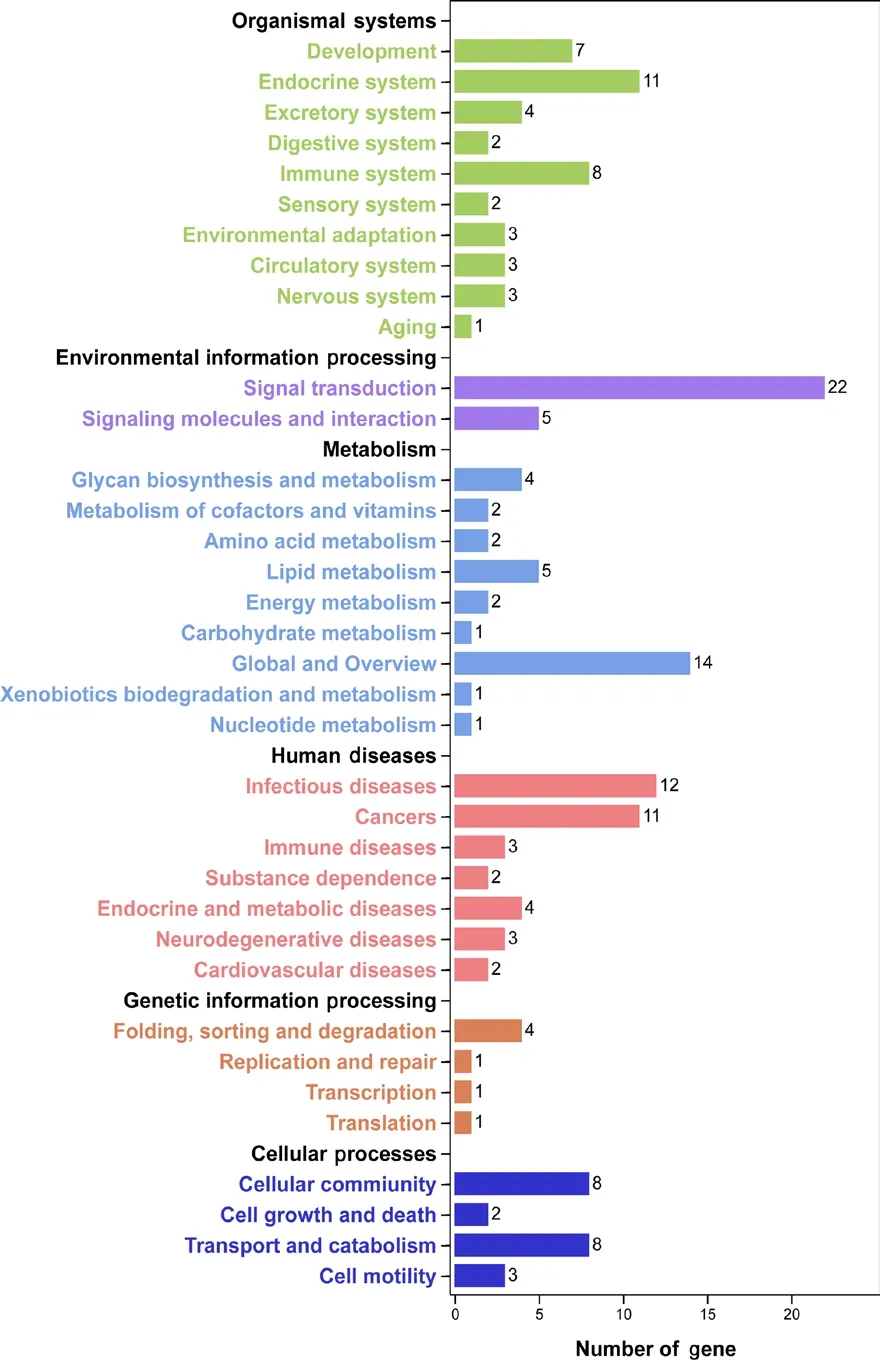
Figure 3 Enriched Kyoto Enrichment of Genes and Genomes (KEGG)pathways of differentially expressed genes in matrix metalloproteinase 7-induced Schwann cells.
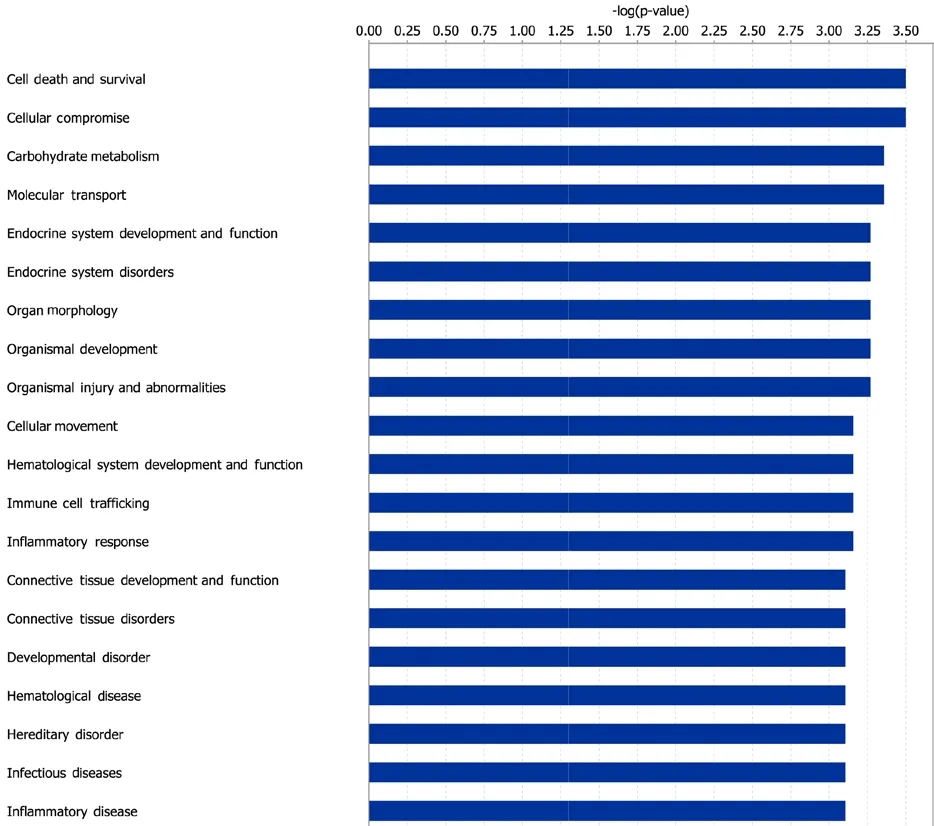
Figure 4 Enriched Ingenuity Pathway Analysis (IPA) diseases and functions.
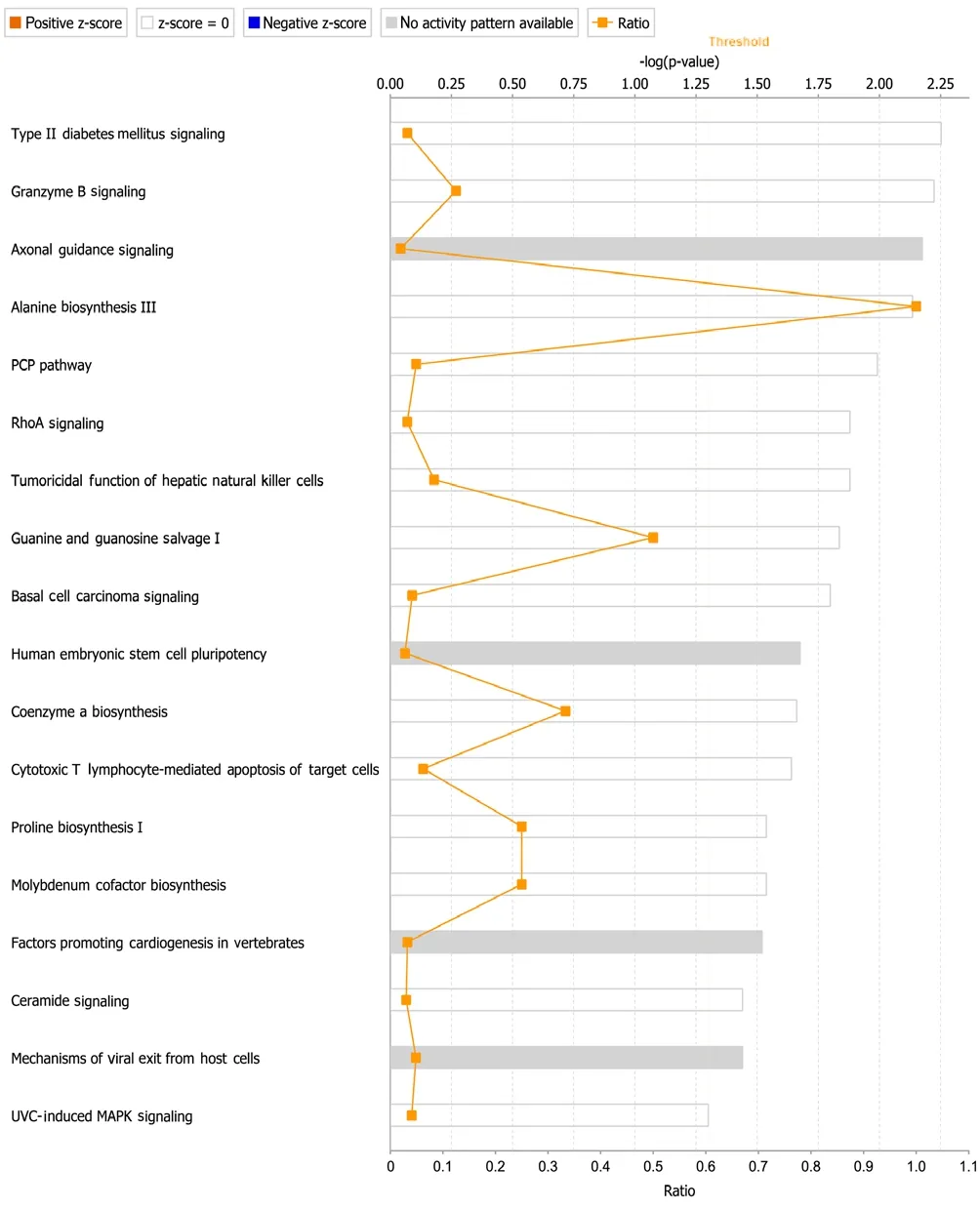
Figure 5 Enriched Ingenuity Pathway Analysis (IPA) canonical signaling pathways.

Figure 6 Overlap of enriched Ingenuity Pathway Analysis (IPA)canonical pathways.
Discussion
RNA sequencing is an advanced high-throughput data analysis technique capable of directly measuring the expression of a large number of RNAs, including coding and non-coding RNAs (Wang et al., 2017, 2018; Wallach et al.,2020). RNA expression reads obtained from this sequencing technique have low noise, high reproducibility, and wide dynamic range (Nagalakshmi et al., 2010). Therefore, sequencing has been widely used in biological fields to investigate the genetic expression of many different systems, such as the peripheral nerve (Yi et al., 2015; Gong et al., 2016; Zhao and Yi, 2019). Sequencing outcomes of nerve stumps post-crush injury demonstrated that many MMPs, especially MMP7 and MMP12, were robustly differentially expressed (Qin et al., 2016). Subsequent functional evaluation demonstrated the important effects of MMP7 in nerve regeneration by exposing SCs to MMP7 (Wang et al., 2019). Furthermore,genetic changes in SCs after MMP7 exposure were examined by RNA sequencing of MMP7-treated or untreated SCs(Wang et al., 2019).
The study of underlying mechanisms, however, was very preliminary and incomprehensive because only 34 genes were found to be significantly changed in expression between MMP7-treated and untreated SCs (Wang et al., 2019).With that in mind, in the current study, we set a relatively lower threshold value, screened a total of 149 MMP7-induced differentially expressed genes in SCs, and re-analyzed these MMP7-induced differentially expressed genes. Consistent with analysis outcomes of our previous study (Wang et al., 2019), our current results indicated the significant involvement of metabolism and biosynthesis-related bioactivities and signaling pathways.
Moreover, by examining a larger number of altered genes,we observed altered expression levels of many extracellular-related genes, includingPsmb8,LOC24906,C1qtnf2,LOC498222,Ndnf,Hsp90aa1,Frmd4b,Rspo3,Egflam,Cklf,Atp6v0a4,Krt19,Prom2,Dpep1,Wnt11,Cep55,Serpinb6b,Syt4,Sema3f,Pcdh12,Efna1,Hap1n3,Apo19a,Cdnf,Rp19,Upk1b,LOC685067,Was,Lingo3,Bmp8b,LOC100911545,Hist1h1d, andRGD1309808. GO cellular component terms“extracellular region”, “extracellular region part”, and “extracellular matrix” were enriched, indicating that MMP7 may conduct its proteolytic effect by degrading extracellular matrix components and cell surface proteins, thus modulating the extracellular matrix of SCs. IPA analysis also demonstrated that “cellular movement” was among the most enriched IPA diseases and functions. A detailed analysis of all IPA biological functions revealed the direct involvement of many cellular movement-related genes, includingNlrp10,S1pr1,Was,Mylk2,Sncb,Stx7,Syt4,A2m,Sema3f,Efna1,Cklf,Lpar2,Prkaa2,Prkcg,Gfra3,Adgrf5,Card10,Gzmb,Hic1,Krt19,Piwil4,Pla2g16,Runx3,Senp1,Sstr5,Wnt11,andMn1.
IPA canonical pathway and pathway overlap analyses further revealed the critical canonical signaling pathways in MMP7-mediated changes, especially axonal guidance signaling. Many genes involved in axonal guidance signaling, includingEfna,Sema3,Dcc,Pak,Wasp,Pkc,Wnt, andBmp, were found to be upregulated or downregulated in MMP7-treated SCs. The directional extension and elongation of axons from neurons to their synaptic targets is essential for the regeneration and functional recovery of injured nerves (Athamneh and Suter, 2015; Suter and Jaworski, 2019;Liu et al., 2020). Receptors located at the leading edge of the axonal growth cone sense attractive and repulsive guidance cues that guide axonal extension and elongation (Gujar et al.,2017; Goulart et al., 2018; Kim et al., 2019). Here, we found that the expression levels of some genes coding for major guidance cues and receptors, such asEfna,Sema3, andDcc,were altered after MMP7 exposure, suggesting that MMP7 may regulate axonal guidance and target reinnervation.
The results of our analysis revealed that MMP7 plays an important role in the regulation of biosynthesis, metabolism,cell movement, axon guidance, and other aspects in addition to acting on extracellular components. However, based on comprehensive and preliminary displays of our results, key pathways and molecules involved in the effects of MMP7 on nerve regeneration need further screening and validation.Taken together, combined use of many bioinformatic tools(including GO, KEGG pathway, and IPA network analyses)for investigation of MMP7-elicited genetic changes in SCs allowed us to obtain a better understanding of the potential involvement MMPs in nerve repair and regeneration. Moreover, our findings provide an experimental and theoretical foundation for the discovery of new therapeutic targets and clinical approaches.
Author contributions:Experiment conception and design: HKW; experiments implementation and data analysis: PJL, GW, XDC; reagents/materials/analysis preparation: HKW, PZ; manuscript writing: HKW. All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare no competing interests.
Financial support:This work was supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions of China[PAPD]. The funder had no roles in the study design, conduction of experiment, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional review board statement:The study was ethically approved by the Laboratory Animal Ethics Committee of Nantong University, China (approval No. 20190225-004) on February 27, 2019.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional files:Additional Figure 1:Identification of primary Schwann cells by immunohistochemistry.Additional Table 1:Full list of differentially expressed genes.
- 中国神经再生研究(英文版)的其它文章
- The role of the TrkB-T1 receptor in the neurotrophin-4/5 antagonism of brain-derived neurotrophic factor on corticostriatal synaptic transmission
- Could non-invasive brain-stimulation prevent neuronal degeneration upon ion channel re-distribution and ion accumulation after demyelination?
- The role of exercise in brain DNA damage
- Combined effect of repetitive transcranial magnetic stimulation and physical exercise on cortical plasticity
- Should mast cells be considered therapeutic targets in multiple sclerosis?
- Neuroprotection mediated by natural products and their chemical derivatives