
Nogo-A and the regulation of neurotransmitter receptors
2020-04-29 02:12BorLuenTang
中国神经再生研究(英文版) 2020年11期
Nogo-A is known to restrict plasticity in the adult central nervous system, and signalling through its cognate receptors modulates synaptic spine architecture and excitatory glutamate transmission via restricting synaptic glutamate receptor levels and their delivery to the post-synaptic compartments. A recent report now indicates that Nogo-A, signaling through Sphingosine-1-Phosphate Receptor 2, also strengthens inhibitory gamma amino acid butyric acid (GABA)ergic transmission by limiting the diffusion dynamics of GABAAreceptors. This reciprocal modulation of excitatory and inhibitory signaling via neurotransmitter receptor dynamics by Nogo-A likely plays important pathophysiological roles in synaptic plasticity during development and injury.
Nogo-A, encoded by theRTN4gene in mammals, is a member of the reticulon family of proteins with a rather unusual membrane association and topology. Although largely found in the endoplasmic reticulum (ER)and has roles in modulating the morphology and functions of the ER(Teng and Tang, 2008), cell surface expressed Nogo-A is a well-known myelin-associated inhibitor of axonal regeneration during central nervous system (CNS) injury. Expressed in both glia and neurons, surface Nogo-A interacts via two separate extracellular domains (Nogo-A-Δ20 and Nogo-66) with several different receptors, namely Nogo receptor 1(NgR1), Sphingosine-1-phosphate receptor 2 (S1PR2), Paired immunoglobulin-like receptor B, as well as Heparan sulfate proteoglycans. In the course of axotomy or die-back upon CNS injury, signaling through these receptors typically activates actin-modifying pathways involving the actin-modulating small GTPase Rho and Rho-associated protein kinase,which generally resulted in growth cone collapse and inhibition of axonal regeneration.
One other important consequence of Nogo-A signaling through its receptors in developing as well as adult neurons is an effect on neuronal architecture, particularly at the synapses. In this regard, signaling through Nogo-A and its receptors modulates dendritic spine morphology and activity-dependent synaptic plasticity (Kellner et al., 2016). Nogo-A could thus modulates learning and memory in regions of the brain such as the cortex (Zemmar et al., 2014) and hippocampus (Zagrebelsky et al., 2017).One way by which this modulation occurs is Nogo-A signaling-mediated changes in glutamate receptor expression and dynamics at excitatory synapses. In this regard, several studies have shown that manipulating Nogo-A-NgR1 signaling could alter synaptic levels, transport, and dendritic spine localization of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPAR) and N-methyl-D-aspartate receptors (NMDAR).
Nogo-A and the modulation of glutamate receptors:The first receptor identified for Nogo-A is the leucine rich repeat-containing, GPI-anchored NgR1. As NgR1 does not span the membrane, signaling is aided by membrane-spanning co-receptors such as the TNF receptor superfamily members p75NTRand TROY, as well as the Leucine-rich repeat and Immunoglobin-like domain-containing protein 1. NgR1 interacts with and is activated by the Nogo-66 domain of Nogo-A, but there is evidence that this interaction may also involve the participation of the N-terminal fragment of Nogo-A. In an early study, silencing of either Nogo-A or NgR1 was shown to increase the levels of the post-synaptic density protein PSD95, as well as AMPAR (GluA1/GluA2) and NMDAR(GluN1/GluN2A/GluN2B) subunits (Peng et al., 2011). Given that these manipulations resulted in the activation of mechanistic target of rapamycin (mTOR), and that rapamycin inhibition of mTOR signaling effectively prevented the up-regulation in glutamate receptor subunits,Nogo-A-NgR1 signaling likely suppresses AMPAR and NMDAR levels via mTOR signaling. A study from Zagrebelsky’s group have also shown that Nogo-A signaling rapidly modulated (within minutes) the spine actin cytoskeleton of hippocampal CA3 pyramidal neurons in slice cultures(Kellner et al., 2016). Indeed, Nogo-A function-blocking antibodies transiently increases F-actin stability and results in an increase in dendritic spine density and length. Nogo-A in adult neurons also appear to restrict AMPAR insertion into hippocampal synaptic sites.
In NgR1-deficient mice, whisker experience-driven synaptic AMPA receptor insertion at the barrel cortex, which is normally completed at around 2 weeks after birth, could continue well into adulthood (Jitsuki et al., 2016). Furthermore, a higher level of AMPARs could be detected on the surface of dendritic spines in the adult barrel cortex of NgR1-deficient mice compared to wild-type mice. In fact, whisker stimulation produced new spines in the barrel cortex of NgR1-deficient but not in those of wild-type adult mice, and these newly formed spines expressed AMPAR at their surfaces. Nogo-A signaling in wild-type mice thus appears to limit synaptic plasticity by restricting the dendritic spine surface delivery of AMPAR. The gene encoding Nogo-A,RTN4, is a known schizophrenia risk gene. In a recent report that examined Nogo-A functions in mouse CA3 hippocampal circuitry using slice cultures,RTN4-/-mice exhibited a hyperactive network (Berry et al., 2018). Interestingly, mGlu3 metabotropic glutamate receptors were downregulated specifically in the CA3 area. TheseRTN4-/-mice also exhibited disordered theta oscillations reminiscent of the phenotype ofGRM3-/-mice. In modified Morris water maze tasks, mice lacking Nogo-A exhibited behavioural deviations that suggests a phenotypic link between Nogo-A and mGlu3 receptors deficiency in schizophrenia. Taken together, the findings summarized above indicate that Nogo-A and its signaling, which in some cases shown to be through NgR1, is important for restraining excitatory transmission by limiting both glutamate receptor levels and synaptic spine insertion.
Nogo-A and the modulation of GABA receptors (GABARs):Given Nogo-A signaling’s influence on glutamate receptors at excitatory synapses, one might anticipate that Nogo-A signaling could also influence inhibitory signaling by GABARs. This was borne out by the finding that NgR1-silencing increased the levels of the metabotropic GABABreceptor 1 (GABABR1) and GABABR2 protein levels but not their transcripts,and without affecting the expression of the ionotrophic GABAAreceptor(Murthy et al., 2013). NgR1 deficiency also resulted in downregulation of the G protein-coupled inwardly rectifying potassium channel (GIRK1).As per the glutamate receptors, in these cases mTOR inhibition by rapamycin also prevented the upregulation of GABABR proteins. Importantly,the increase in GABABRs and GIRK1 levels corresponded to an increased expression of these proteins at the plasma membrane. Analysis of synaptosomes from the hippocampal tissues of NgR1 knockout mice also showed an increased in GABABR2 and GIRK levels. It thus appears that Nogo-A-NgR1 could also affect inhibitory synaptic transmission through modulation of the G-protein-coupled GABABRs and GIRK1.
A new receptor for Nogo-A entered the field in 2014, when Schwab’s laboratory identified S1PR2, a cognate receptor for the signaling lipid molecule Sphingosine-1-Phosphate (S1P), as a functional Nogo-A receptor that could act in repressing synaptic plasticity (Kempf et al., 2014).S1PR2 is a G-protein coupled receptor, and unlike the Nogo-66-interacting NgR1, binds to Nogo-A-Δ20 at a site distinct from the S1P binding pocket. S1PR2 signals via the G protein Gα13, and activates RhoA though the Leukemia-associated Rho Guanine nucleotide exchange factor(LARG). Blocking S1PR2 signaling strongly enhances long-term potentiation in the hippocampus and motor cortex of wild-type mice, but not inRTN4-/-animals.
In hippocampal neurons, inhibitory GABAergic synaptic strength is defined by the number of postsynaptic chloride-selective ionotropic GABAAreceptors (GABAARs). In particular, fast GABAergic inhibition is regulated through the control of GABAAR diffusion. In a recent report,Zagrebelsky et al. (2017) have now shown that Nogo-A signaling through S1PR2 restricts synaptic GABAAR diffusion. The authors found that synaptic localization of Nogo-A is regulated in an activity-dependent manner. Blocking Nogo-A signaling through S1PR2 using function-blocking antibodies specific for the Nogo-A-Δ20 domain reduced receptor number at synaptic sites, and this corresponded to a reduction in the amplitude of GABAergic miniature inhibitory post-synaptic currents at CA3 hippocampal neurons. The apparent increase in GABAAR diffusion rate is associated with an increase in calcium transients with the loss of Nogo-A function, which is not observed with NgR1 silencing. This also requires dephosphorylation of the GABAAR γ2 subunit at Ser327 by the Ca2+-dependent phosphatase calcineurin. Importantly, a gain-of-function Δ20 inhibitory peptide suppressed the calcium transients. Nogo-A signaling through S1PR2 therefore strengthens inhibitory GABAergic transmission by restricting the diffusion of synaptic GABAAR, likely by suppressing calcium transients resulting from neuronal activity (Fricke et al., 2019).Notably, this contrasted with how Nogo-A signaling regulates excitatory transmission, which is effected via the restriction of glutamate receptor levels, as well as synaptic transport and insertion.
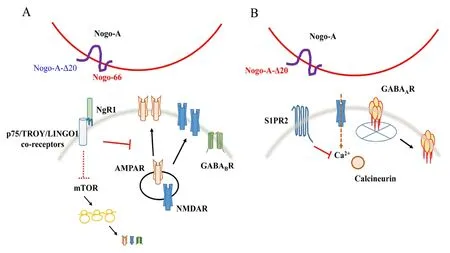
Figure 1 Schematic diagram illustrating the role of Nogo-A signaling in modulating neurotransmitter receptors at synapses.
Implications and perspectives:The findings outlined above illustrated a potentially generalizable principle by which Nogo-A signaling modulates synaptic plasticity in the adult CNS. By reciprocal regulation of glutamate receptors and GABARs at their respective post-synaptic compartments,Nogo-A curbs excitatory neurotransmission and stabilizes inhibitory ones. Conversely, a reduction or loss of synaptic Nogo-A would strengthen excitatory synaptic transmission while reducing inhibitory synaptic transmission. Although most of the work thus far is done with hippocampal slices and tissues, this reciprocal mode of Nogo-A modulation of synaptic transmission likely also occurs in the synapses of other brain regions in different guises. Both Nogo-A and S1PR2 are widely expressed in the brain, including the cortex, cerebellum and spinal regions (Kempf et al., 2017). Nogo-A over-expression in the presynaptic boutons of cerebellar Purkinje cells of transgenic mice resulted in the destabilization of GABAergic synapses and a compensatory upregulation of postsynaptic GABAARs (Aloy et al., 2006). Nogo-A’s inhibitory transmission modulatory effect also appears to be dependent on the type of Nogo-A receptor present.
An interesting point to note here is that the GABAARs responded rather specifically to loss of Nogo-A signaling through S1PR2, as an earlier report of NgR1 manipulation did not affect GABAAR expression(Murthy et al., 2013), and NgR1-silencing did not elicit the calcium transients associated with S1PR2’s effect on GABAAR (Fricke et al., 2019). The deciphered mechanism implies that Nogo-A signaling through different receptors could differentially regulate the expression and dynamics of excitatory and inhibitory neurotransmitter receptors (Figure 1). How this signaling differentiation occurs is not particularly clear at the moment.Nogo-A-NgR1 signaling activates Rho-associated protein kinase, and Nogo-A-S1PR2 signaling would likewise activate Rho via the Rho GEF LARG. In hippocampal neurons, fast GABAergic inhibition is activity-dependent, and primarily regulated by NMDAR-mediated calcium influx. Glutamate signaling of NMDARs and the resulting sustained calcium influx increase GABAARs lateral diffusion via Ca2+-activated calcineurin’s dephosphorylation of GABAARs’ γ2 subunit at Ser327. As observed, Nogo-A-S1PR2 signaling but not Nogo-A-NgR1 signaling affected calcium transients that altered GABAAR stabilization, and localization of Nogo-A at synapses is rapidly reduced upon an increase in neuronal activity (Fricke et al., 2019). Nogo-A-S1PR2 signaling could therefore suppress activity-induced calcium transients. In this regard, how Nogo-A levels change with activity remains to be investigated. S1P’s binding to S1PR2 is known to result in calcium release from intracellular stores as well as extracellular calcium influx. However, deciphering exactly how Nogo-A’s engagement of S1PR2 result in a suppression of calcium influx would require further work.
Another point to note is that the role of S1PR2 in Nogo-A’s modulation of GABAAR dynamics and GABAergic transmission were shown by function-blocking antibodies and the soluble Δ20 inhibitory peptide,which actions are acute or pharmacological in nature. It would be interesting to see if similar changes occurs with S1PR2 silenced slices, and how GABAAR dynamics are altered in S1PR2 knockout, or brain conditional knockout animals. Finally, what implications would the findings discussed here have in terms of Nogo-A signaling-based interventions during CNS injury? Schwab’s laboratory has recently shown that vascular repair and network formation in a mice stroke model is improved in Nogo-A or S1PR2 knockout mice, or wild-type mice infused with a function-blocking Nogo-A-Δ20 antibody (Rust et al., 2019). These results supports the notion that attenuation of Nogo-A-S1PR2 signaling, with attenuation of excitatory synaptic transmission and stabilization of inhibitory transmission, would be therapeutically beneficial to ischemic CNS injuries.
BLT was supported by the NUS Graduate School for Integrative Sciences and Engineering.
Bor Luen Tang*
Department of Biochemistry, Yong Loo Lin School of Medicine;National University of Singapore Graduate School for Integrative Sciences and Engineering, National University of Singapore,Singapore
*Correspondence to:Bor Luen Tang, PhD, bchtbl@nus.edu.sg.
orcid:0000-0002-1925-636X (Bor Luen Tang)
Received:December 30, 2019
Peer review started:January 10, 2020
Accepted:March 10, 2020
Published online:May 11, 2020
doi:10.4103/1673-5374.282250
Copyright license agreement:The Copyright License Agreement has been signed by the author before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Hans Rudolf Widmer, University Hospital and University of Bern, Switzerland; Marta Zagrebelsky, Technical University Braunschweig, Germany.
Additional file:Open peer review reports 1 and 2.
- 中国神经再生研究(英文版)的其它文章
- The role of the TrkB-T1 receptor in the neurotrophin-4/5 antagonism of brain-derived neurotrophic factor on corticostriatal synaptic transmission
- Could non-invasive brain-stimulation prevent neuronal degeneration upon ion channel re-distribution and ion accumulation after demyelination?
- The role of exercise in brain DNA damage
- Combined effect of repetitive transcranial magnetic stimulation and physical exercise on cortical plasticity
- Should mast cells be considered therapeutic targets in multiple sclerosis?
- Neuroprotection mediated by natural products and their chemical derivatives