
B2—FeAl合金基本性能及间隙缺陷的理论研究
2020-04-22 12:40蓝春香唐帆斌
中国金属通报 2020年3期
蓝春香,唐帆斌
(南宁学院 通识教育学院,广西 南宁 530000)
B2-FeAl合金具有高熔点、质地轻、低成本、较强的耐磨性和抗氧化性等一系列优良特点,成为中高温腐蚀环境中备受关注的结构材料之一[1]。当合金材料所处环境温度高于0K时,完整晶格受热振动作用发生不同程度的晶格畸变,在晶内形成各类错综复杂的晶体缺陷。
常见的点缺陷的存在对合金材料的电阻率、导热性及力学性能和机械性质的影响极大,亦成为材料研究的热点。最初由Daw和Baskes[2,3]基于准原子近似和密度范函理论建立的半经验式理论模型(Embedded Atom Method,EAM原子嵌入势),掀起金属材料关于体性质、晶界、表面、合金和热物理性质等方向的理论研究的热潮。
Fu等[4]使用局域密度泛函理论研究了B2-FeAl的平衡点缺陷,认为富Fe合金中Fe原子占据Al原子晶格形成Fe反位缺陷,富Al合金中Al原子占据Fe原子晶格形成Al反位缺陷。Fahnle等[5]用统计力学结合第一原理方法研究了B2-FeAl点缺陷和原子扩散。Bessen等[6]构造了包含非中心项的半经验EAM多体势广泛地研究了FeAl合金的体性质、点缺陷和热学性质。Vallhe等[7]用EAM模型研究了B2-FeAl点缺陷、位错核心结构和层错等缺陷性质。Bakke等[8]用扩展的Miedema半经验模型方法研究了B2-FeAl的点缺陷形成能。
本文将借助欧阳义芳提出的改进型EAM原子嵌入势[9],结合分子动力学模拟方法从原子尺度上研究B2-FeAl金属间化合物的基本物理性能及合金体相中含B原子的点缺陷的性质。
1 模拟方法
利用MS,以初始晶格常数为a=2.889Å、简单CsCl结构建立一个10a×10a×10a,由2000个原子构成的B2-FeAl原胞。在零压、周期性边界条件下充分的弛豫以获得平衡态下合金的晶格常数a、结合能Ec、熔点Tm、弹性系数等力学参量,再与实验值和其他理论值对比,以验证EAM势函数选取的可靠性。
在确定势函数的可靠性后,以势函数对应的平衡晶格常数重新构建一个由2000个原子组成的B2-FeAl原胞,用以研究原胞内存在B原子时点缺陷的性质。将单个半径r=0.95Å的B原子放在原胞中(基于原胞结构的周期对称性,体系内任何位置上的单原子缺陷都是等价的),分别置换Fe位原子、Al位原子形成反位缺陷BFe、BAl,或在原胞内与近邻的Al、Fe原子形成等原子距的四面体间隙BT(由2Al+2Fe组成)及Al、Fe原子分别作为最近邻原子的八面体间隙BO-Al(2Al+4Fe)、BO-Fe(2Fe+4Al),共五种缺陷类型。
Yan[10]在研究硼原子在B2-FeAl[100(]010)表面发生刃型位错时指出,由不同原子构成的合金体系,原子间的相互作用对研究缺陷的形成能不能忽略,必须考虑构成体系的各类原子的化学势。在无杂质条件下,可通过对B2-FeAl原胞构造出空位及反位四种缺陷,利用MVT系宗分别求出Fe原子和Al原子的化学
考虑B2-FeAl块体中含单个B原子的缺陷形成时,缺陷形成能的计算分别如式(1)、(2)所示:
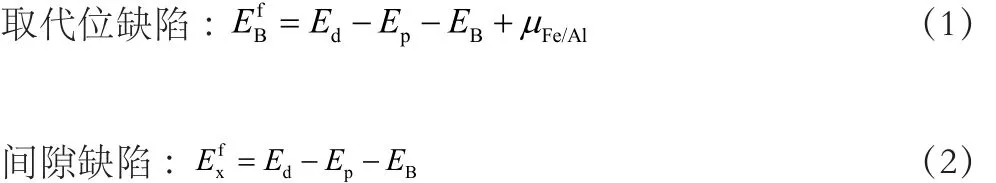
2 结果与讨论
2.1 晶格常数的变化
晶体的内聚能起伏表征晶体结构的稳定性,与温度有关,体系内聚能越低,对应的结构越稳定,即为平衡状态。同一晶体结构在不同温度下充分弛豫会发生热膨胀,晶胞的晶格常数会发生改变。将FeAl原胞置于适宽的温度范围内进行弛豫,当达到其收敛条件时, 我们就能得到晶格常数下对应晶胞系统的总能量值。
图1为能量-晶格参数变化曲线图。从图中可知FeAl合金的最低内聚能为4.250eV,对应的平衡晶格常数a=2.981Å。计算结果与Shu[11]利用EAM势通过分子动力学模拟得到的结果相同,略大于Villars[12]的实验值和Fu[13]DDF理论的第一性原理计算。
2.2 合金的熔点
将FeAl原胞在0.0002Ps 步长条件下从1K 升温到3000K的熔化过程进行动力学模拟。在体系温度逐渐上升至相变之前,体系的总能量与温度呈线性关系。当系统升温至合金熔点附近时,由于合金发生相变熔化潜热释放导致能量突变,可认为能量突变的温度为熔点。如图2所示,即Tm=2588.67K ,接近实验值[12]。
2.3 弹性常数与力学性能
固体材料的弹性常数、体积模量、剪切模量及杨氏模量可以提供材料力学性能方面的性质,是表征材料刚度和稳定性的重要信息。立方晶系中的三个弹性常数是相互独立的,可通过弹性常数矩阵Cij[14]。
在外部压力为0GPa时,根据EAM模型对优化后的新模型进行模拟得到合金的三个弹性常数分别为208.9GPa、140.7GPa、106.9Gpa,与Nye[15]的实验值及Shu[11]的分析改进型EAM势分子动力学模拟结果及Huber[14]通过分子光谱分析拟合的结果接近,稍小于Fu[13]第一性原理模拟结果。
对于立方晶系,体积模量、剪切模量和杨氏模量B、GE可通过Voigt-Reuss-Hil(lVRH)近似获得,计算结果分别为:162.3GPa、76.5GPa、201.4GPa。
根据Pugh准则,体积模量B和剪切模量G的比值B/G可以作为判断材料是脆性或者延性的条件。如果B/G大于1.75,则这种材料为延性,否则为脆性。文中B/G=2.12,说明FeAl合金具有一定的延展性。
2.4 点缺陷性质
为了获得体系平衡状态下的最小能量,胞内所有原子力场之和不大于10-4eV/Å。T=0K时,根据Yan[10]的计算模型求出体系内Fe、Al、B的化学势分别为 :µFe=-4.50eV 、µAl=-3.79eV 、µB=-5.31eV ,与实验值[16]较好的吻合。结合(1)、(2)缺陷形成能计算公式,B2-FeAl合金中BFe、BAl、BT、BO-Al、BO-Fe五种缺陷形成能分别为:0.84eV、0.57eV、1.45eV、1.56eV、1.25eV。本文计算结果与Yan[10]、Raulo[17]的研究结果接近。三种不同方法计算都得到B取代Al位的缺陷形成能最低,这说明B杂质原子在B2-FeAl块体中易于形成Al反位缺陷,其次是Fe反位缺陷,最后是间隙缺陷。
3 结论
结合EAM模型与分子动力学对B2相FeAl合金的计算与分析,可以得出如下结论:

图1 FeAl不同晶格常数对应的结合能
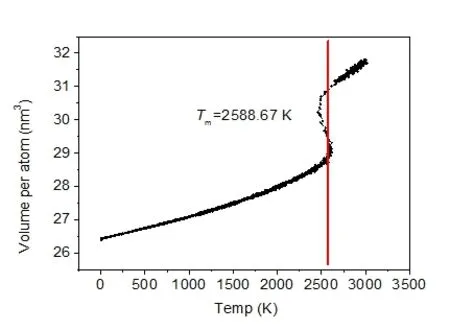
图2 升温时FeAl总体积随温度的变化
(1)通过EAM势计算FeAl合金的基本性能所得结构与实验值和其他理论值相符较好,说明采用EAM势研究FeAl合金是可靠的。
(2)在B2-FeAl合金中,B原子形成反晶格位缺陷比形成间隙缺陷更容易,占据Al原子位的缺陷形成能最低。
猜你喜欢
太原理工大学学报(2022年5期)2022-09-23
光子学报(2022年6期)2022-07-27
当代作家(2021年11期)2021-12-17
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
电子制作(2018年12期)2018-08-01
卷宗(2018年14期)2018-06-29
新高考·高一物理(2016年3期)2016-05-18
云南中医学院学报(2012年3期)2012-07-31
