
MoTe2的电子结构及光学性质的理论研究
2020-04-15 01:59:40马睿华刘珊珊周慧颖任梦琦伍冬兰
井冈山大学学报(自然科学版) 2020年1期
马睿华,刘珊珊,辛 霞,周慧颖,任梦琦,伍冬兰
MoTe2的电子结构及光学性质的理论研究
马睿华,刘珊珊,辛 霞,周慧颖,任梦琦,*伍冬兰
(井冈山大学数理学院,江西,吉安 343009)
利用密度泛函理论第一性原理方法对MoTe2的能带结构、能态密度和光学性质进行了理论计算,得到能带结构、态密度、光吸收谱、能量损失谱和介电函数等光学性质。结果表明:MoTe2具有间接带隙宽度为1.066 eV的半导体材料,价带主要由Mo的5s4p价电子和Te的5s5p价电子起主要作用;导带由Mo和Te的4d价电子起主要作用。由获得的光学性质可知,介电函数的实部和虚部的峰值都出现在低能区;位于可见到紫外区域的光子具有很强的吸收,最大吸收系数为2.84×105cm-1;同时在光子能量为16.40 eV处出现了共振现象,其它区域内电子之间共振非常微弱。这些光学性质奠定了该材料在制作微电子和光电子器件方面的作用。
MoTe2;电子结构;光学性质;第一性原理
0 引言
近年来,随着半导体器件理论和制造技术的发展,由最初的硅材料到以石墨烯为主的层状纳米材料[1-2],再到过渡金属硫族化合物材料的发展中,其中二维过渡金属硫族化合物(TMDs)[3]由于其较高的载流子迁移率、适当的带隙、较大的开关比以及带隙具有层数依赖性等优点,迅速成为材料领域的研究热点[4-6]。二碲化钼(MoTe2)是TMDs家族重要的组成部分,其除了具有上述提到的优异性质外,还具有一些十分独特的性质。与其它TMDs材料相比,MoTe2的能带隙最小(1.1 eV)[7],并且MoTe2是唯一一种半导体的三棱柱结构(2H相)和金属性质的扭曲八面体结构(1T’相)均能稳定存在的材料,而且这两种相结构在一定条件下可以发生可逆转变[8]。这些优异的性质使MoTe2材料在电子和光电器件领域具有广泛的应用前景。
然而尽管MoTe2材料具有众多优异的性质,该材料的制备却是一个很大的难题,目前化学气相沉积法(CVD)被认为是制备二维MoTe2薄膜最有效的方法,但是由于Mo、Te之间电负差很小,导致Mo和Te原子间通过化学反应形成化学键很困难,因此不容易得到纯相的MoTe2薄膜[9]。本文利用密度泛函理论第一性原理的超软赝势的方法进行理论计算,分析了MoTe2的能带结构、态密度和光学性质等物理特性,这为制备该材料提供理论参数参考。首先利用基于密度泛函方法的从头计算量子力学程序软件CASTEP[10]对晶体的结构进行优化,计算分析得到了能带结构图、态密度图以及介电函数图等示意图,再根据优化后的结构图进一步分析得到MoTe2各物理性质参数,再利用相关数据分析计算出MoTe2的光学性质。
1 理论计算方法
1.1 模型及计算方法
图1是MoTe2的分子结构示意图,从图中可以得到MoTe2晶体结构属于类似于石墨烯的六方晶系中的正交晶系。晶格常数为a=0.352 nm;b=0.352 nm;c=1.397 nm。
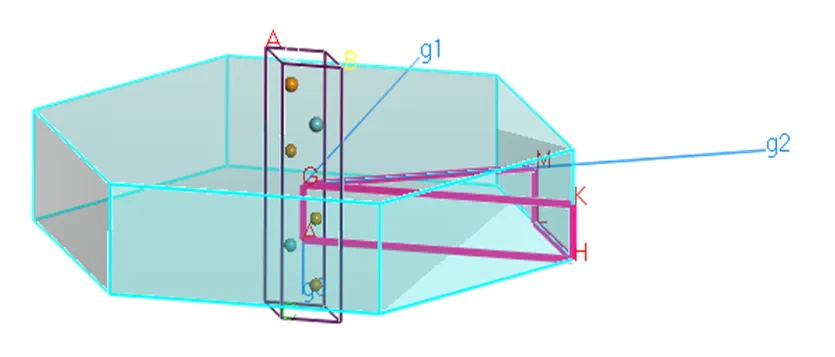
图1 MoTe2晶体结构示意图
本文基于密度泛函理论的从头计算量子力学程序软件CASTEP程序包,采用第一性原理中的超软赝势的方法理论模拟晶体、晶体界面和晶体表面的特性。利用BFGS算法优化MoTe2晶体结构,计算分析能带结构、电子态密度和光学性质。计算过程中,忽略电子的自旋影响,采用超软赝势平面波的方法处理离子及价电子之间的相互作用,其中截断能为Ecut=320 eV,价电子选取Mo的4d55s1电子和Te的5s25p4组态电子,其它的轨道电子固定为芯电子。自洽精度为每一个原子的能量不超过5×10-7eV,布里渊区的k点设置为8×8×2。
1.2 MoTe2的光学性质
MoTe2的光学特性函数可以由复介电函数()1()+2()[11]转换得到。其中复介电函数和复折射率关系如下:

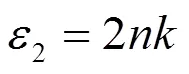
式中为消光系数,代表折射率。从MoTe2跃迁的概率导出介电函数的虚部,再根据科拉莫斯-克勒尼之间的色散关系,由介电函数虚部计算进而得到函数实部,关系如下[12]:
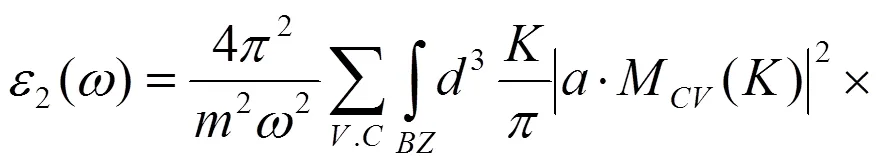
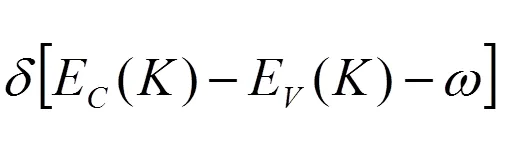
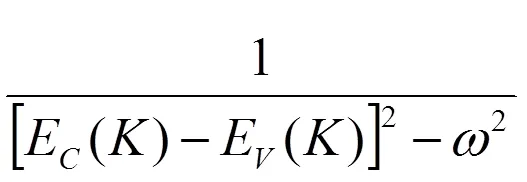
由于折射率、吸收谱和反射系数等光学性质的函数与介电函数存在一定的联系,因此可以通过KK关系得出其他光学参量。
2 计算结果及讨论
2.1 MoTe2的电子结构
图2是MoTe2电子的能带结构图。图中表明MoTe2导带的最小值为1.036 eV,在第一布里渊区的K点取得,在第一布里渊区的G点取得价带的最大值为-0.03 eV。由于最大和最小值的取得点不在同一点上,说明带隙结构为间接带隙,带隙宽度为Egap=1.066 eV,与实验值1.1 eV[7]吻合较好。各布里渊区中高对称点的导带和价带能量特征值如表1所示。根据晶体场论配位对中心离子的d轨道和f轨道的影响,利用分子轨道理论对双原子分子的分子结构及配位场进行了有效的近似,可以影响费米面附近的能量。分析能带结构可得轨道分裂发生在G点,是因为Mo的d轨道电子之间存在扰动的缘故,致使导带分裂变成导带及次导带。从图中还能够看出,G点位置出现了简并能峰,这是因为价带和次价带在G点出现了简并现象。综上分析可知,能带结构构成的缘由,是由于电子在分裂d轨道上的再次分布引起能级分裂形成的。

图2 能带结构图

表1 MoTe2各K点在价带最高点Ev和导带最低点Ec的能量值
图3是MoTe2电子态密度分布图,直观地表示了各电子态对导带与价带的作用。由总态密度分布图可知,各峰值出现的能量区间有-61.394 eV~-60.297 eV,-35.65eV~-34.335 eV,-13.265eV~-9.879 eV,-6.116~0.28 eV和0.28 eV~8.558 eV。结合Mo和Te原子的分态密度图进行分析可知:当MoTe2处于价带-61.394 eV~-60.297 eV的能量区间时,峰值主要由Mo原子的5s电子和Te原子的5s电子共同决定的;而处于-35.65 eV~-34.335 eV能量区间时,由Mo的4p电子和Te原子的5p电子共同决定的;当处于-13.265 eV~-9.879 eV的能量区间时,主要由Te的5s电子决定,其它电子贡献较少;当处于-6.116~0.28 eV能量区间时,主要由Te的4d5p电子和Mo的4d电子共同决定,其余电子在此能量区间的贡献较小;而能量区间在0.28 eV~8.558 eV导带能量范围的中,主要由Mo的4d电子和Te的4d5p电子共同决定的,Te的5s电子和Mo的4p5s电子只有少量贡献。结合分态密度图可推出,中心原子的轨道容易排列组合,形成杂化轨道。从图3中还可以发现,当Mo的4d电子和Te的5s5p电子的能量值为0.28 eV时将形成杂化轨道,这个杂化轨道形成了价带顶。从能带结构图中可以知道简并度出现在价带的G点,这种现象也可以被证明。从分态密度图3(b)可以看出,在1.597 eV能量值附近,Mo的4d电子的次峰表明导带主要由Mo的4d态决定,而结合能带结构图表明它恰好对应于导带的最小值并构成导带的底部,并且在过度自由状态下的Mo的4d电子很可能在影响下形成扰动态Te的5p电子,这是由于自旋轨道耦合会诱导轨道分裂形成的,与能级结构分析结果一致。
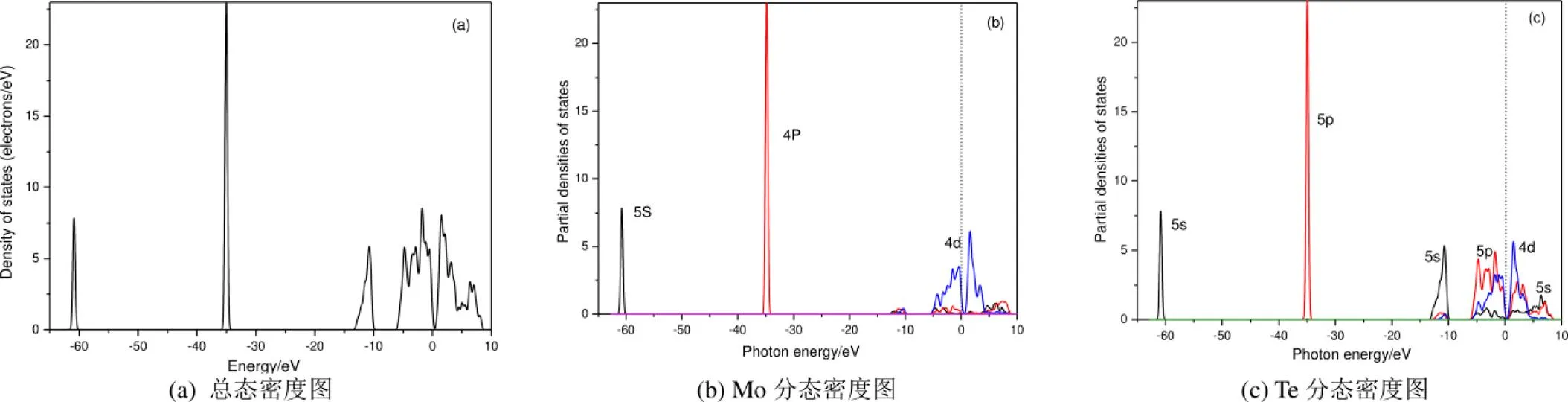
图3 态密度图
2.2 MoTe2的光学性质
2.2.1 MoTe2的复介电函数
由于介电函数的虚部能够通过一定的关系得出其他的光学性质,而且介电函数的虚部及实部有着因果关系,他们之间可以通过KK关系相互转化得到,因此介电函数在光学性质的计算过程中是非常重要的一部分。
图4为入射光子能量范围0~20 eV内的介电函数,表明介电函数的实部Rm和虚部Im随光子能量的变化关系。图中表明实部的极大值为25.73 eV,而虚部的极大值为21.12 eV,1.90eV和16.33 eV两个能量为实部的曲线和实轴的交点,介电函数的实部取得负值时的能量范围是3.6~16.33 eV,实部的极小值为-7.03 eV而虚部的极小值为0 eV。介电函数虚部Im反映物质对光的吸收情况,一般由图中的介电峰呈现,因为介电峰是由价带和导带之间的电子相互转变形成的,从这个介电峰值,还可以看出电子结构和其他光学性质及光谱信息。将介电函数图和态密度图进行对比分析,可以得出电子的跃迁的信息,再通过计算能级之间的能量差,则可推出介电常数虚部的谱线频率,然后依靠能量之间存在的差异,计算出介电常数虚部Im及其谱线频率,最后由电子的跃迁概率推导出强度。
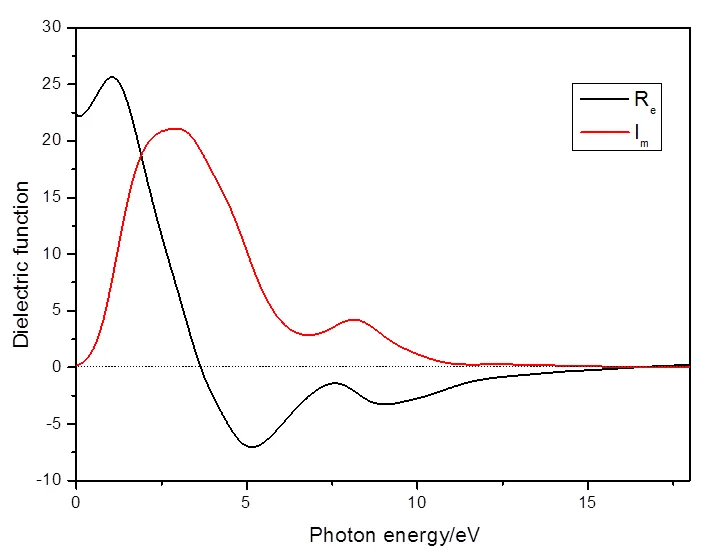
图4 复介电函数
2.2.2 MoTe2的复折射率
根据介电函数分析得到的信息,由于介电函数的虚部与其它光学性质存在一定的相互关系,因此可以通过获得的介电函数的虚部参数,获得该材料的折射率、吸收谱和能量损函数等光学性质。图5为MoTe2的复折射率随频率的分布关系图,和分别为折射率和消光系数。分析图5可知,光电子能量在0~1.14 eV范围内出现了最大的折射率,但当大于1.14 eV时,折射率逐渐变小,且在7.87 eV时,出现一个较小的峰值,最后逐渐降低为0。由复折射率与介电函数之间的关系可得消光系数,如下:
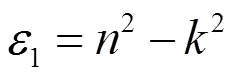

由图可知,消光系数在= 4.53 eV处有一极大值,在< 0.01 eV及> 16.58 eV区域内,趋于零,但在曲线的递增和递减区,曲线出现峰值和低谷,它们的位置分别为6.48 eV和7.87 eV。

图5 MoTe2的复折射率
2.2.3 MoTe2的反射谱
图6为随光子能量变化时反射率的变化趋势。如图6所示,反射率出现了三个峰值和两个低谷的变化趋势,在0~5.82 eV的能量范围内,反射率具有0.64 eV的峰值;在5.82~7.6 eV,反射率具有0.37 eV的低谷;在7.6~11.21 eV,反射率具有0.82 eV的最大峰值;在11.21~14.60 eV,反射率具有0.69 eV的低谷;在14.60~16.09 eV,反射率具有0.82 eV最大的峰值,之后迅速减少趋于零。这些分析表明反射率大部分值都大于0.4,说明MoTe2具有相对较高的反射率,处于该区域内的入射光大部分会被反射回来,具备较强的反射性质。通过比较分析发现,最大反射率0.82 eV小于MoS2(0.93 eV)[13],说明MoTe2晶体更易吸收光子。

图6 MoTe2的反射谱
2.2.4 MoTe2的吸收谱
通过介电函数虚部与吸收系数之间的关系,可得出吸收系数如图7所示。

关系式中,为介电函数的虚部,n为折射率,c为真空中的光速,ω为入射光的频率。从MoTe2的吸收光谱图可看出,吸收峰分别位于5.20 eV和9.05 eV。这是因为Te的5s电子跃迁到Mo的4d电子和Mo的4 s电子跃迁到Te的5 p电子态。同时吸收光谱在高能区域更高,且在9.05 eV处存在一个最大吸收峰283651 cm-1(MoS2的值为317027 cm-1)[13],这说明MoS2晶体具有更强的吸收红外光子的特性,这与反射率分析结果一致。当光子能量大于17.53 eV时,MoTe2的吸收值趋于零,这种现象称为“透明现象”,意味着当红外光通过MoTe2晶体时,不会被吸收。
2.2.5 MoTe2的能量损失函数
物质的能量损失函数揭示了各状态的粒子持续集体震荡的关系特性。根据MoTe2的能量损失函数分别与复介电函数实部和虚部所对应的关系式即可推出能量损失函数如图8。由图可知,在能量E=16.40 eV处,函数存在最大值为63.39 eV,之后曲线由峰值近乎笔直减小直到0。结合态密度和能带结构图可推出:在E=16.40 eV处Mo的4s、4d和Te的5p电子发生共振,处在原子最活跃的时候。在E<13.18 eV和E>17.26 eV范围内发生的能量损失值几乎为0,也就是说电子之间的共振非常微弱,即电子之间没有发生共振现象。通过比较分析发现,最大能量损失值63.39 eV小于MoS2(139.01 eV)[13],说明该晶体的粒子持续集体震荡特性小于MoS2晶体。
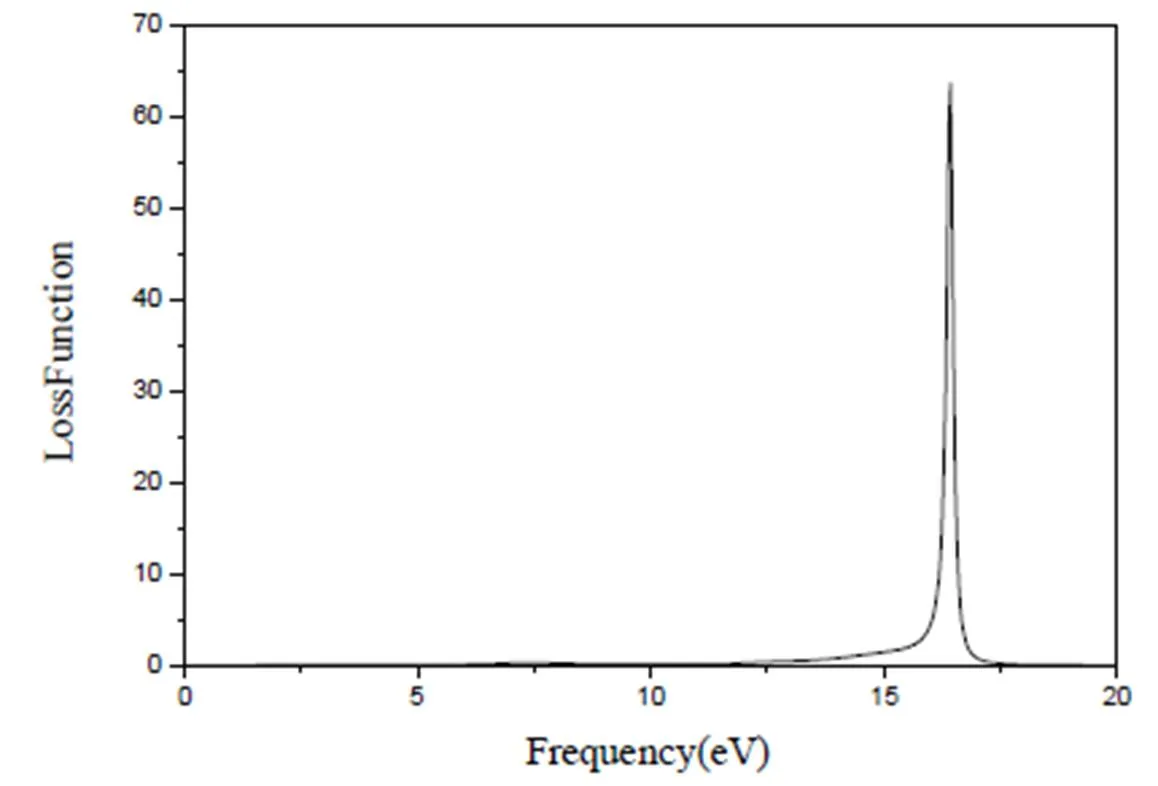
图8 MoTe2的能量损失函数
2.2.6 MoTe2的光电导率
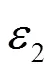
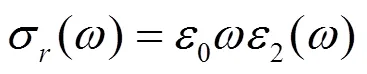
关系式中σr为光电导率的实部,ε0为真空介电场数,ε0为介电函数的虚部。物质的光电导率的实部代表能带间电子跃迁信息,峰值为诸多电子从价带到导带跃迁的贡献之和。图9中Im为光电导率虚部,Re为光电导率实部。其中Re曲线出现两个峰值,分别位于3.62 eV和8.25 eV位置处,分析发现这两个峰值恰好与介电函数的虚部和吸收谱的峰值的位置相差不远。Im曲线中的峰值位于实部递减区域,谷位出现在实部的递增区域,其峰值分别在5.36 eV和9.29 eV处。不难看出,该位置与介电函数的虚部以及吸收谱的峰值位置非常相近。
3 结论
本文采用第一性原理的超软赝势方法计算和分析了MoTe2的能带结构、态密度和光学性质。得出材料MoTe2具有间接带隙宽为1.066 eV的半导体材料,价带和导带均由Mo和Te的价电子起作用形成的。由光学性质分析得出介电函数的实部和虚部的峰值都出现在低能区;光吸收谱显示材料MoTe2对紫外光的吸收作用最大,吸收系数为2.84×105cm-1,其它区域吸收系数趋于0,出现了“透明现象”;光子能量位于16.40 eV位置附件出现了共振现象,其它位置的电子之间共振非常微弱。这些电子结构和光学性质奠定了该材料在制作微电子和光电子器件方面的主导作用,尤其是在紫外探测器应用方面有着潜在的应用前景。
[1] Govinda R B, Rao H S S R, Rao C N R. Decoration of few-layer graphene-like MoS2and MoSe2by noble metal nanoparticles[J]. Journal of Cluster Science, 2012, 23(3): 929-937.
[2] Mondal B, Sengupta K, Rana A, et al. Cobalt corrole catalyst for efficient Hydrogen evolution reaction from H2O under ambient conditions: reactivity, spectroscopy, and density functional theory calculations[J]. Inorganic chemistry, 2013, 52(6): 3381-3387.
[3] Wlison J A, Yoffe A D. Transition metal dichalcogenides discussion and interpretation of observed optical, electrical and structural properties[J]. Adv. Phys,1969, 18: 193-335.
[4] Zhang X, Tan Q H, Wu J B, et al. Review on the Raman spectroscopy of different types of layered materials[J]. Nanoscale, 2016, 8: 6435-6450.
[5] Luxa J, Jankovsky O, Sedmidubsky D, et al. Origin of exotic ferromagnetic be havior in exfoliated layered transition metal dichalcogenides MoS2and WS2[J]. Nanoscale, 2015, 8: 1960-1967.
[6] Chen X. Optical study on two dimensional transition metal dichalcogenides[J]. University of Hong Kong, 2015, 44: 2629-2642.
[7] Xu X A, Li X B, Feng Q L, et al.Thermodynamics and kinetics synergetic phase-engineering of CVD grown single crystal MoTe2films[J].Crystal Growth & Design, 2018,18(5): 2844-2850.
[8] Zhou L, Xu K, Zubair A, et al. Large-area synthesis of high-quality uniform few-layer MoTe2[J]. Journal of the American Chemical Society, 2015, 137: 11892-11895.
[9] Wang Y, Xiao J, Zhu H, et al. Structral phase transition in monolayer MoTe2driven by electrostatic doping[J]. Nature, 2017, 550: 487-491.
[10] Pu J, Yomogida Y, Liu K K, et al. Highly flexible MoS2thin-film transistors with ion gel dielectrics[J]. Nano Letters, 2012, 12: 4013-4017.
[11] 沈学础. 半导体光谱和光学性质[M].北京: 科学出版社, 2002: 76-94.
[12] Kang D F, Zhou Y Z, Yi W, et al. Superconductivity emerging from a suppressed large magnetoresistant state in tungsten ditelluride[J]. Nature communications, 2015, 6:392-397.
[13] 范哲梅,蒋立鹏,伍冬兰. 第一性原理研究MoS2的电子结构和光学性质[J]. 井冈山大学学报:自然科学版, 2019, 39(6): 7-13.
STUDYON THE ELECTRIC STRUCTURE AND OPTICAL ELECTRICAL CHARACTERISTIC OF MoTe2
MA Rui-hua, LIU Shan-shan, XIN Xia, ZHOU Hui-ying, REN Meng-qi,*WU Dong-lan
(School of Mathematic and Physical, Jinggangshan University, Ji’anJiangxi 343009, China)
In this paper, the first-principle of ultra-soft pseudopotential method of density functional theory is used to theoretically calculate the energy band structure, energy density and optical properties of MoTe2. The results show that MoTe2is a semiconductor material with an indirect band gap and the bandgap width is1.066eV. The formation of the valence band and the conduction band is caused by the valence electrons of Mo and Te. Specifically, the formation of the valence bands by the 5s4p states of Mo and the 5s5p states of Te play different roles, the conduction band formation, the 4dstate of Mo and Te have a greater effect, other states has only a little effect. Through the calculation and analysis, the optical properties of MoTe2can be obtained: dielectric function, complex refractive index, energy loss function and so on. It is found that MoTe2has a strong absorption of visible photons in the ultraviolet region with a maximum absorption coefficient of 2.84×105cm-1. MoTe2has a resonance phenomenon at about 16.40eV, and the resonance between electrons is very weakin other regions. The optical properties of MoTe2lay a foundation for its role in the fabrication of microelectronics and optoelectronic devices, which can provide theoretical reference for further study of MoTe2material in the future.
MoTe2; electronic structure; optical properties; first principles
1674-8085(2020)01-0010-06
O561.3
A
10.3969/j.issn.1674-8085.2020.01.003
2019-10-12;
2019-11-13
国家自然科学基金项目(11564019, 11147158);江西省教育厅科研课题(GJJ170654)
马睿华(1999-),男,河南洛阳人,井冈山大学数理学院本科生(E-mail:maruihua@jgsu.edu.cn);
刘珊珊(1999-),女,江西九江人,井冈山大学数理学院本科生(E-mail:liushanshan@jgsu.edu.cn);
辛 霞(1999-),女,山东烟台人,井冈山大学数理学院本科生(E-mail:xinxia@jgsu.edu.cn);
周慧颖(1998-),女,山东潍坊人,井冈山大学数理学院本科生(E-mail:zhouhuiying@jgsu.edu.cn);
任梦琦(1998-),女,山东东营人,井冈山大学数理学院本科生(E-mail:renmengqi@jgsu.edu.cn);
*伍冬兰(1978-),女,江西吉安人,教授,博士,主要从事分子结构与光谱研究(E-mail:wudonglan1216@sina.com).
猜你喜欢
吉首大学学报(自然科学版)(2023年6期)2023-12-22 08:18:20
物理学报(2023年20期)2023-11-16 10:43:34
中等数学(2021年6期)2021-08-14 02:35:50
数学学习与研究(2020年23期)2020-01-11 08:47:27
吉首大学学报(自然科学版)(2018年3期)2018-07-03 03:14:12
Chinese Journal of Chemical Engineering(2017年5期)2017-05-28 10:22:54
卷宗(2016年8期)2016-11-15 20:56:37
电测与仪表(2016年2期)2016-04-12 00:24:48
中小企业管理与科技·下旬刊(2015年4期)2015-04-08 12:49:35
物理学报(2010年6期)2010-09-08 06:05:24
