
基于TCGA研究铁死亡相关基因对乳腺癌患者的预后
2020-04-13 03:10侯晨阳胡艳玲黄代政毛宇昂
广西大学学报(自然科学版) 2020年6期
侯晨阳,胡艳玲,黄代政,毛宇昂
(广西医科大学 信息与管理学院,广西 南宁 530000)
0 引言
在全世界女性队列中,乳腺癌是最常见的恶性肿瘤之一,死亡率高居第二位,且发病率和死亡率一直逐年上升,对女性的健康有着极大的隐患[1]。在分子水平上,乳腺癌是一种具有高异质性的肿瘤,按照分子分型,乳腺癌分为四种亚型,即Luminal A型、Luminal B型、HER2过表达型、Basal-like型,且每一种亚型的乳腺癌患者的预后具有明显差异[2]。此外,世界各地不同的地理环境也造成了乳腺癌患者预后和总体生存率具有差异。由于乳腺癌病因复杂,加上高度的异质性,使得探究疾病预后成为一种挑战。另外,乳腺癌治疗手段的局限性的使然,开发新的预后模型成为一种必然。
铁死亡是一种铁依赖性的,区别于细胞凋亡、细胞坏死、细胞自噬的新型的细胞程序性死亡方式,在致命的脂质过氧化驱动下,从而诱导细胞死亡[3-4]。近年来,相对于具有耐药性肿瘤的传统治疗方法,通过铁死亡机制,诱导肿瘤癌细胞死亡成为一种有前景的治疗手段。除了铁死亡的诱导因子外,许多基因也被确定为铁死亡的标志物。然而,这些和铁死亡相关的基因与乳腺癌患者预后的关系还尚不清楚。
在本研究中,首先从公共数据库下载了乳腺癌患者的mRNA表达谱及相应的临床数据。然后,在TCGA队列中通过铁死亡相关的差异表达基因构建了一个多基因预后模型,并在METABRIC队列中验证此模型。最后,笔者进一步进行了功能富集分析,以探讨其潜在的生物学机制。
1 材料与方法
1.1 数据下载
本研究在TCGA(https://portal.gdc.cancer.gov/repository)数据库下载了1 053名病人的乳腺癌mRNA数据及相应的临床信息,数据下载格式为标准化之后的FPKM格式。用于验证的数据来自于乳腺癌国际联盟的分子分类学(METABRIC)数据库(http://www.cbioportal.org/datasets),包括1 904名乳腺癌病人的mRNA-seq数据及其相关的临床信息,此数据的表达矩阵同样是经过标准化之后的数据。METABRIC是一项加拿大与英国合作项目,旨在根据有助于确定最佳治疗过程的分子特征将乳腺肿瘤分类为更多的亚类。TCGA和METABRIC的数据都是公开可获得的。因此,本研究无需获得当地伦理委员会的批准。此外本研究遵守TCGA和METABRIC数据访问和使用准则。
同样,与铁死亡相关的基因来自于铁死亡数据库(FerrDb)(http://www.zhounan.org/ferrdb)的基因标志物和文献检索得到基因的并集,共计148个基因[5-7]。
1.2 预后模型的构建与验证
在TCGA队列中,通过R包“limma”对肿瘤组织和癌旁组织做基因的差异表达分析,筛选出与铁死亡相关的差异基因(DEGs)。筛选阈值为|log2FC|>1,FDR<0.05。同时进行的是对总生存率(overall survival, OS)的单因素Cox回归分析,以筛选具有预后价值的铁死亡相关基因。运用R包“venn”对具有差异和预后价值的铁死亡相关基因取交集,并且运用STRING (version 11.0) 数据库构建蛋白互作网络[8]。为了达到最大化减小过度拟合风险的目的,笔者采用LASSO Cox 回归分析并结合R包“glmnet”构建预后模型[9-10]。回归分析中的自变量为差异和预后交集的基因表达量,因变量是TCGA队列中乳腺癌患者的总体生存率和状态,得出最优模型的基因及对应的回归系数。根据每个基因的标准化表达水平及其相应的回归系数计算每一个患者的风险评分[危险得分=e^(基因1表达量×相应的回归系数+…+基因n表达量×相应的回归系数]。根据得到风险评分求其中位数,将患者分为高风险组和低风险组。在生存分析中,根据高、低风险两组运用R包“survminer”构建生存曲线。同时使用R包“timeROC”进行ROC曲线分析,评估其特征基因预后模型的预测能力。
1.3 功能富集分析
运用R包“limma”对高风险患者组和低风险患者组做基因的差异表达分析,筛选出风险差异基因,筛选阈值为|log2FC|>1,FDR<0.05。通过R “clusterProfiler”对高、低风险两组中的差异基因进行基因本体(gene ontology, GO)、京都基因和基因组百科全书(kyoto encyclopedia of genes and genomics, KEGG)分析。此外,基于R包“gsva”,通过单样本基因集富集分析(ssGSEA)对16个免疫细胞浸润打分,分析13个免疫相关的通路[11]。
1.4 统计学分析
在肿瘤组织和癌旁组织差异分析时采用t检验。高、低风险组间的总体生存率比较采用Kaplan-Meier法。采用单因素和多因素Cox回归分析来确定总体生存期的独立预后因子。所有的数据统计分析均基于R平台(Version 4.0.2)。如果以上分析方法没有进行特殊说明,则认为P<0.05具有统计学意义,且所有P值均为双尾分布。
2 结果
通过筛选可用数据,本研究最终纳入了1 012名来自TCGA队列的乳腺癌患者和1 901名来自METABRIC队列的乳腺癌患者。
2.1 确定与铁死亡相关的差异预后基因
确定TCGA队列中选择的铁死亡相关基因见图1。在肿瘤组织和癌旁组织间做差异表达时,发现大部分与铁死亡相关的基因都存在差异表达(117/148,79.1 %)[图1(a)]。在单因素回归分析中,有20个差异基因与总体生存的预后相关[图1(b~c)]。因此,这20个基因被纳入后续的预后分析,且所有的基因的FDR值均小于0.05。这些基因之间的相互作用网络图表明TXRND1、CS、GPX4、G6PD为中心基因[图1(d)]。此外,图1(e)展示了这些基因之间的相关性。
2.2 构建预后模型
根据前期分析得到的20个基因的表达谱,采用LASSO Cox回归分析构建预后模型。经过回归分析,得出最优的16个基因及其对应的相关系数(图2)。危险评分计算公式为:e^(0.184×CISD1的表达量-0.189×AIFM2的表达量+0.132×ALOX15的表达量+0.196×NCOA4的表达量+0.061×CHAC1的表达量+0.215×CS的表达量-0.055×GPX4的表达量+0.033×SQLE的表达量-0.041×CAPG的表达量+0.179×G6PD的表达量+0.386×NGB的表达量+0.106×GCLC的表达量+0.013×HSBP1的表达量+0.053×PRDX1的表达量-0.167×SLC1A4的表达量+0.036×BNIP3的表达量)。根据危险得分的中位值将患者分为高风险组(506例)和低风险组(506例),如图2(a)。如图2(b)所示,高风险组的早死概率高于低风险组的患者。在进行生存分析时,Kaplan-Meier曲线显示低风险组的存活率明显高于高风险组,且P<0.001,如图2(c)。采用ROC曲线评估总体生存风险评分的预测性能,曲线下面积(AUC)在1 a内为0.65,2 a内为0.70,3 a内为0.67, 如图2(d)。
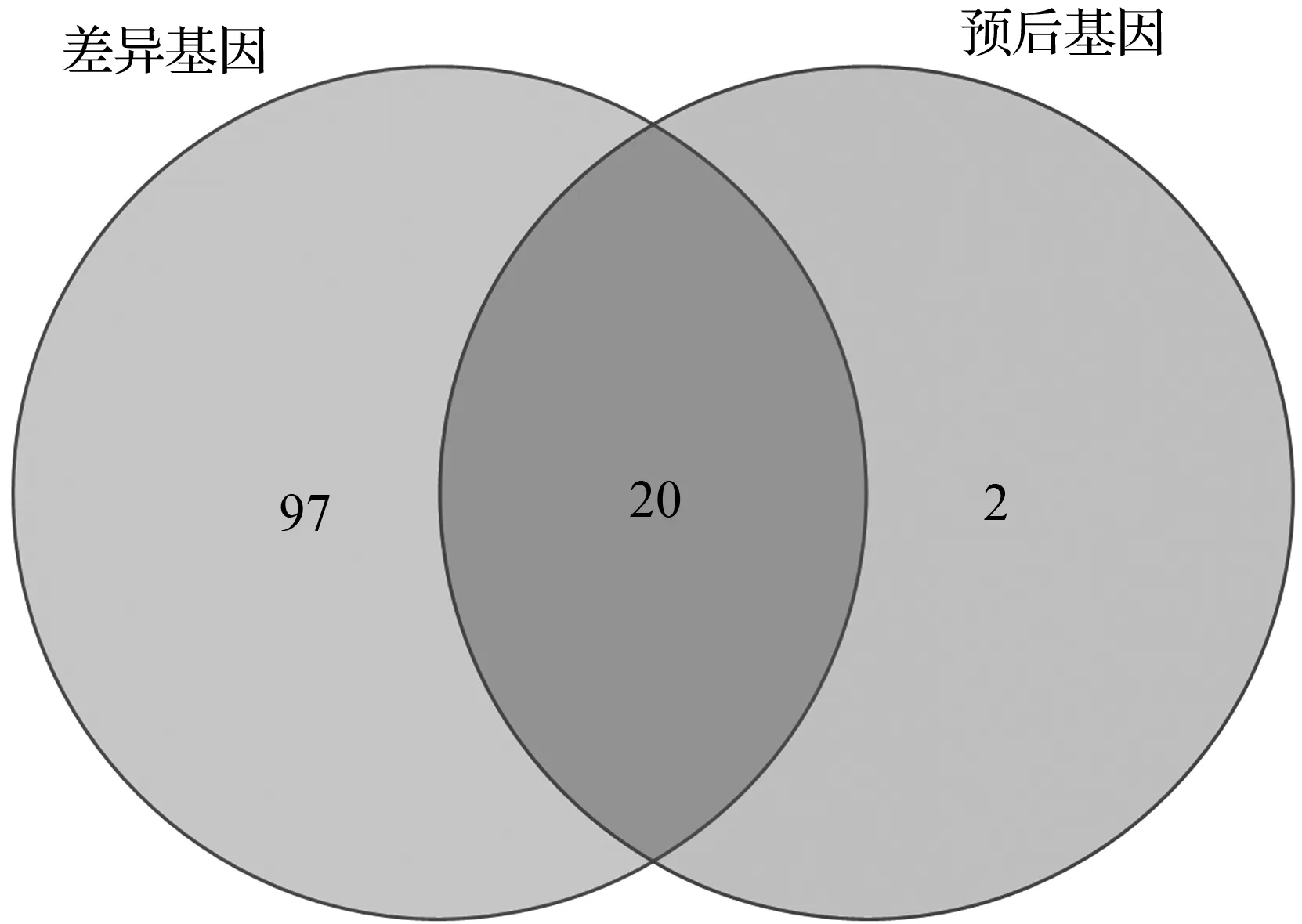
(a) 用维恩图来鉴定肿瘤间差异表达的基因以及与OS相关的邻近正常组织

(b) 20个重叠基因在肿瘤组织中的表达情况
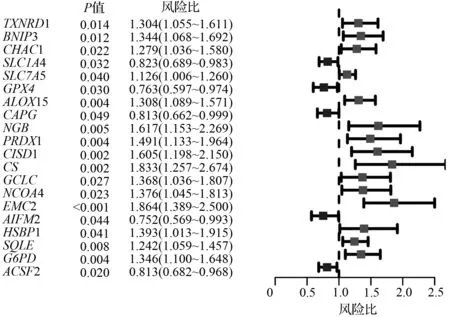
(c) 显示单因素回归分析结果的森林图,包括基因表达与OS的Cox回归分析
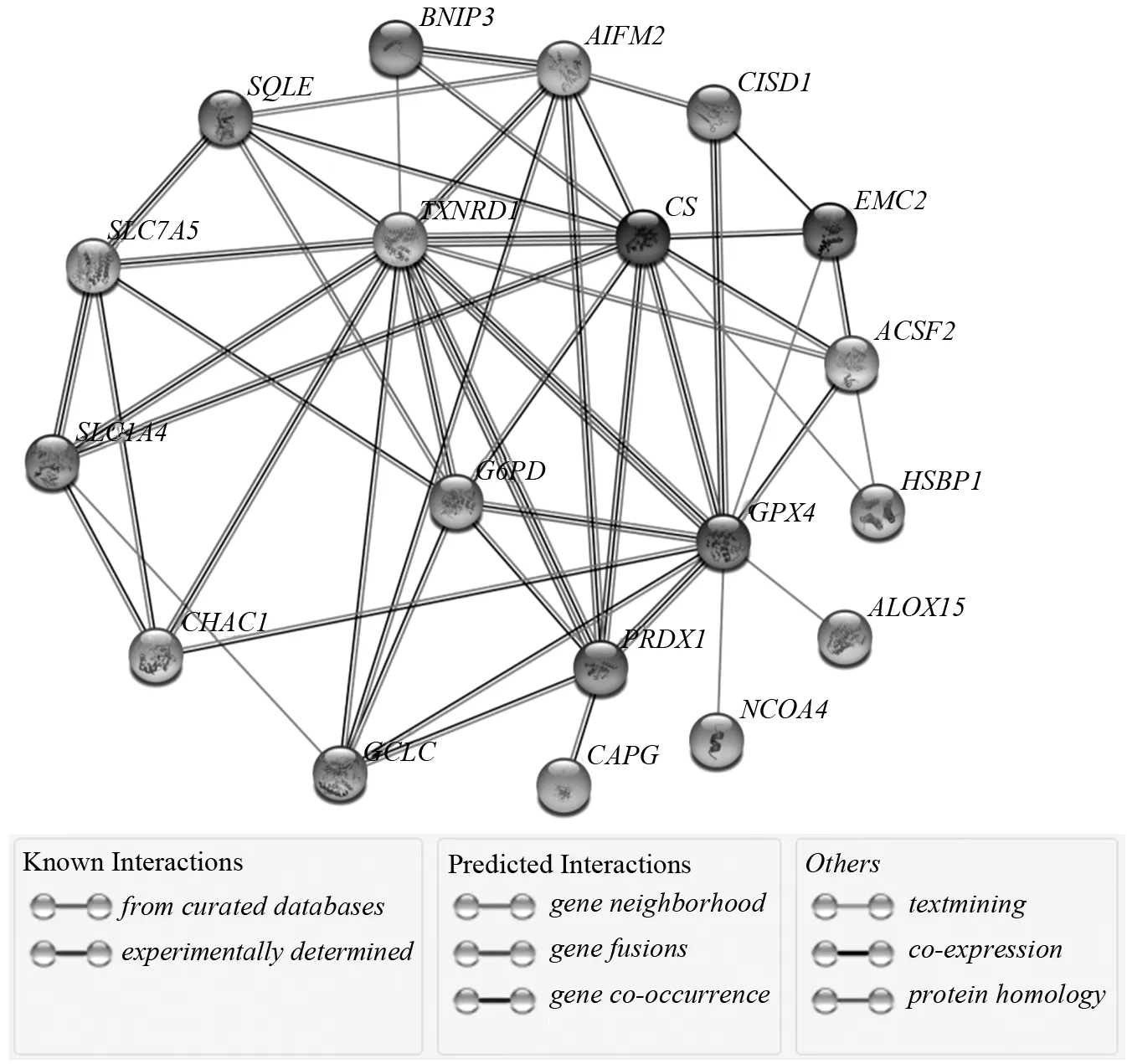
(d) 从STRING数据库下载的基因间互作网络显示了候选基因之间的相互作用
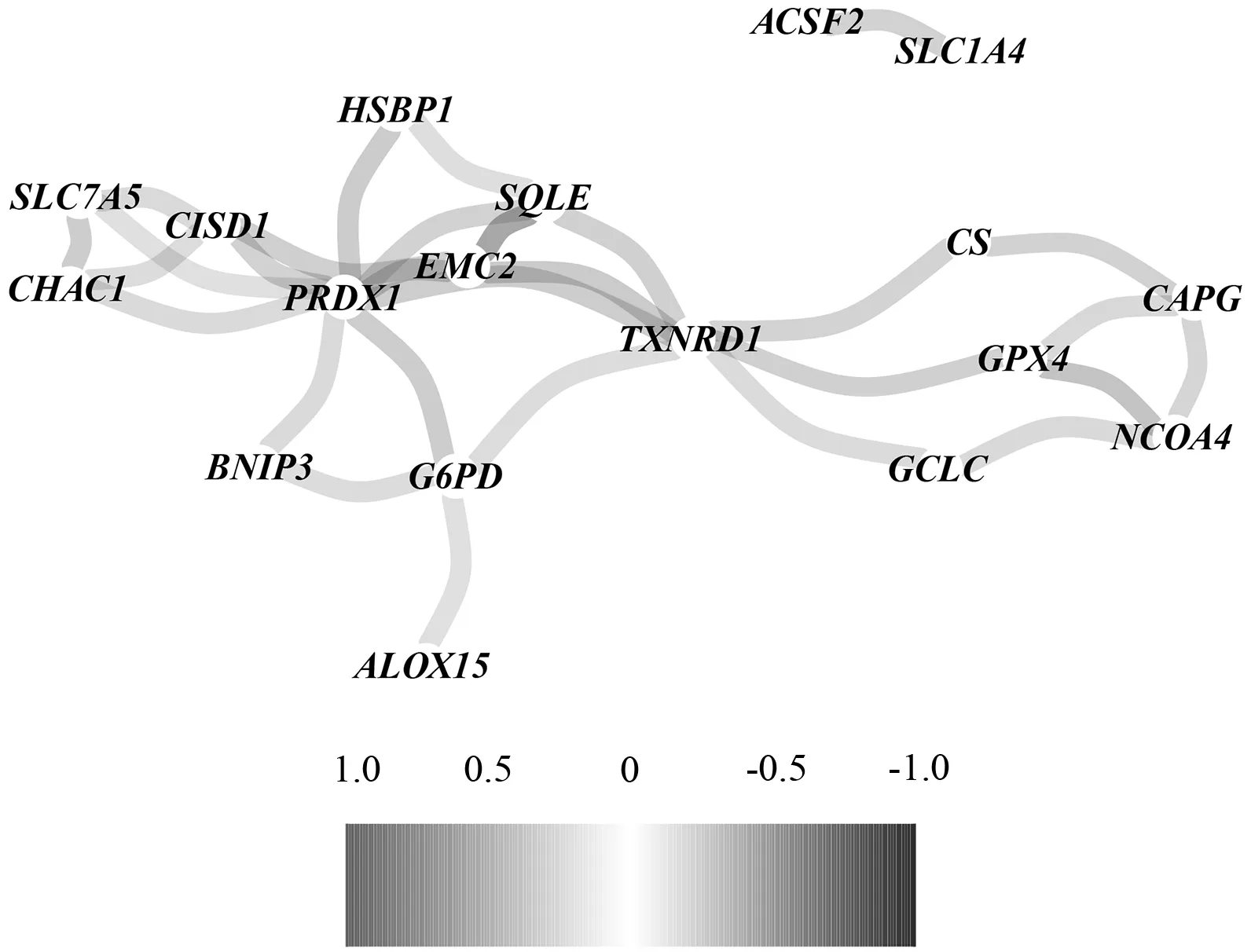
(e) 候选基因的相关网络,相关系数用不同的颜色表示
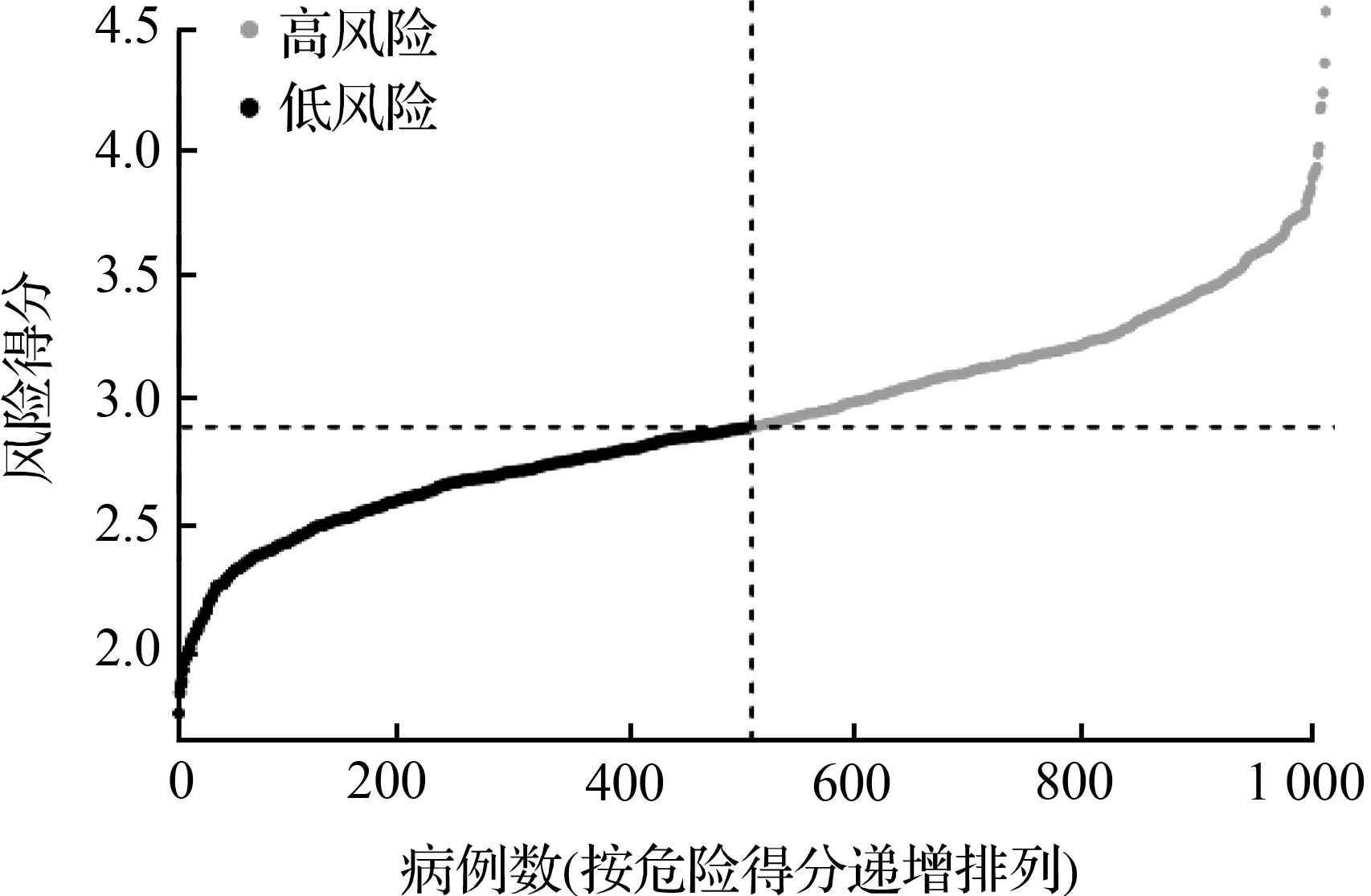
(a) TCGA队列中风险评分的分布和中值
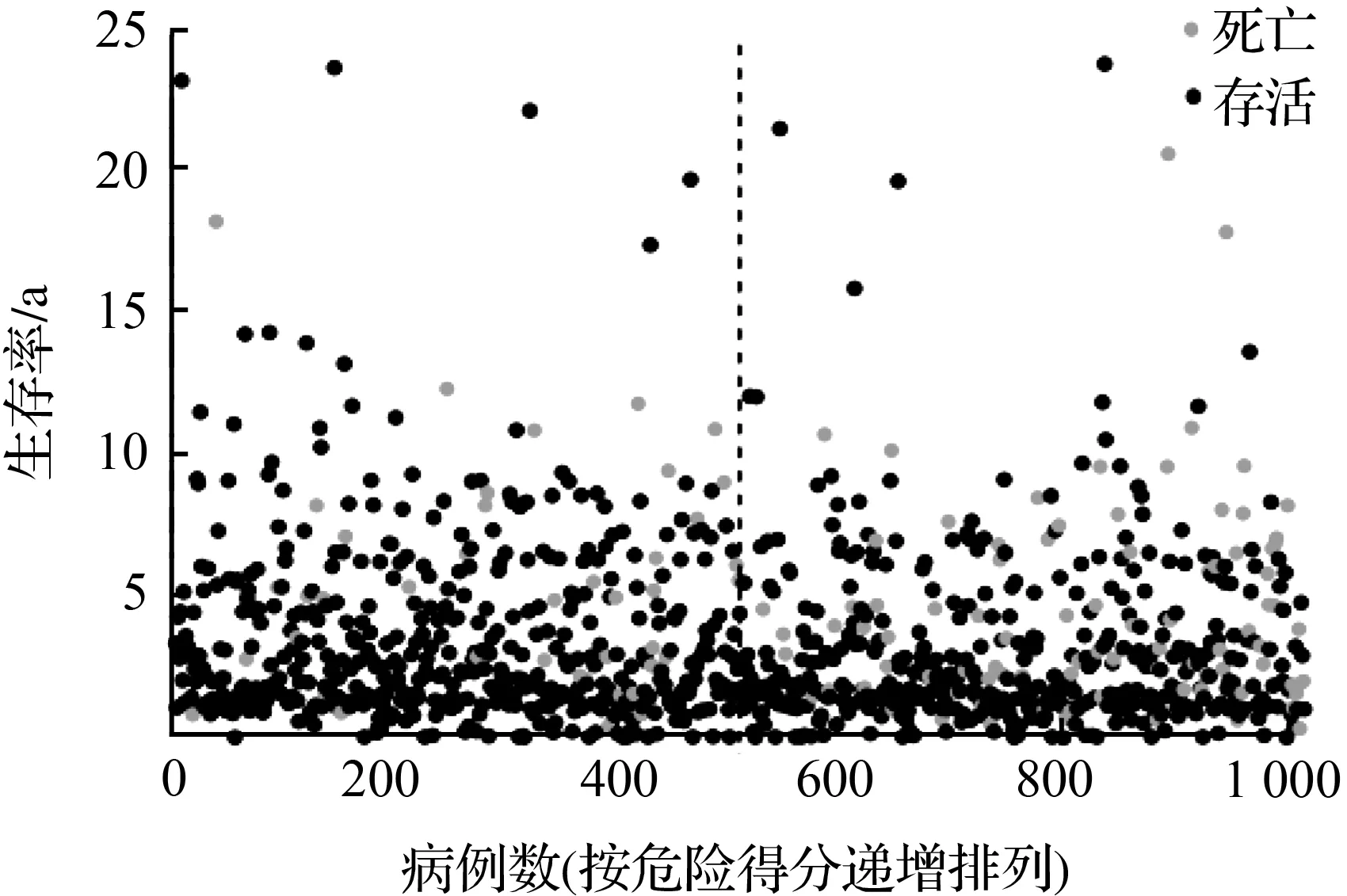
(b) TCGA队列中OS状态、风险评分的分布
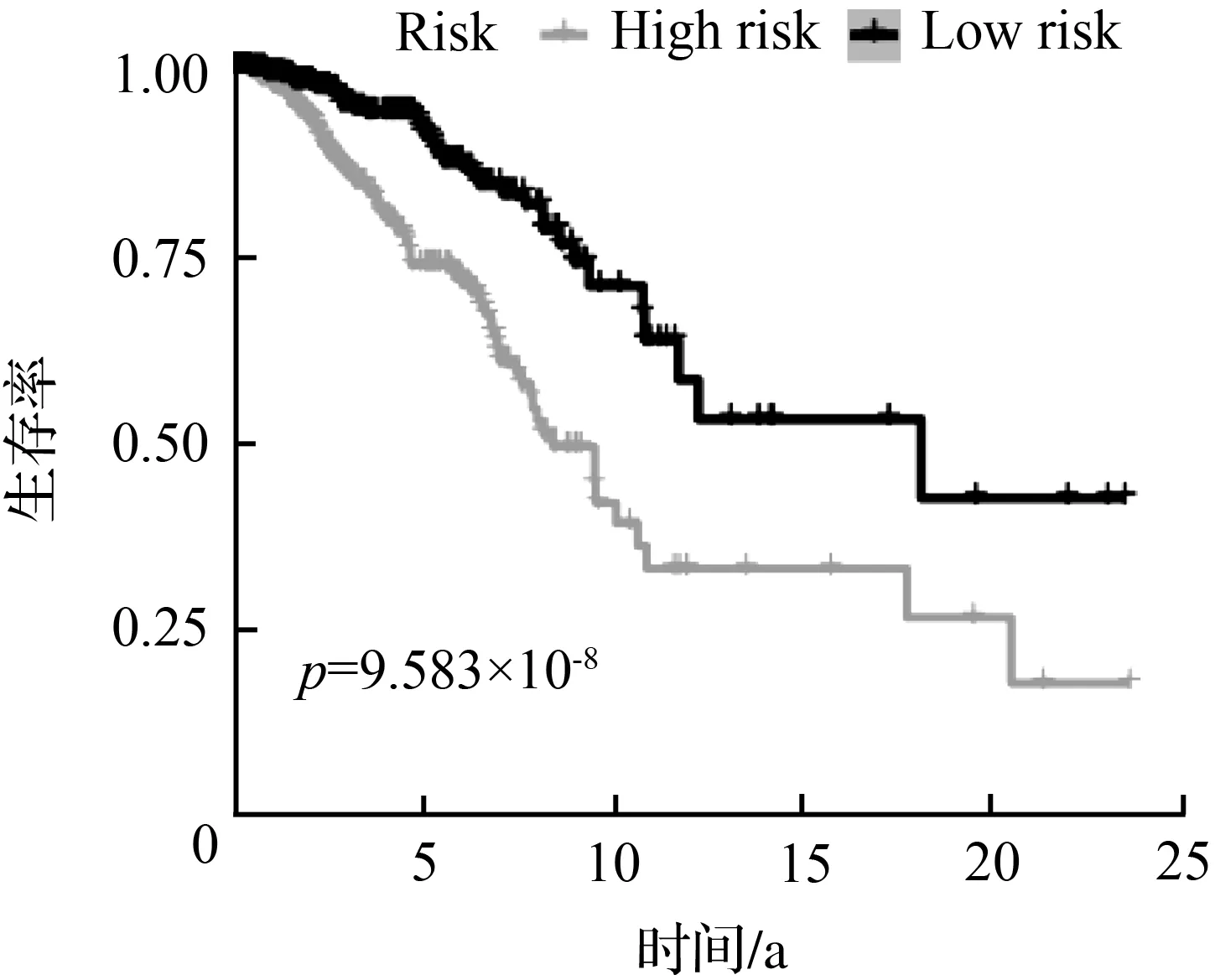
(c) Kaplan-Meier曲线, TCGA队列中高风险组和低风险组患者的生存率

(d) ROC曲线下面积(AUC)评测预后模型
2.3 验证预后模型
为了验证TCGA队列构建预后模型的稳健性,本研究将METABRIC队列的患者按照与TCGA队列相同的危险得分公式进行计算,并将计算的中值分为高风险组和低风险组,如图3(a)。与TCGA结果相似,高风险组的患者早死概率高于低风险组的患者,如图3(b)。同样地,生存曲线显示低风险组的存活率明显高于高风险组,且P<0.001,如图3(c)。此外,ROC曲线显示AUC在1 a内为0.66,2 a内为0.62,3 a内为0.62,如图3(d)。
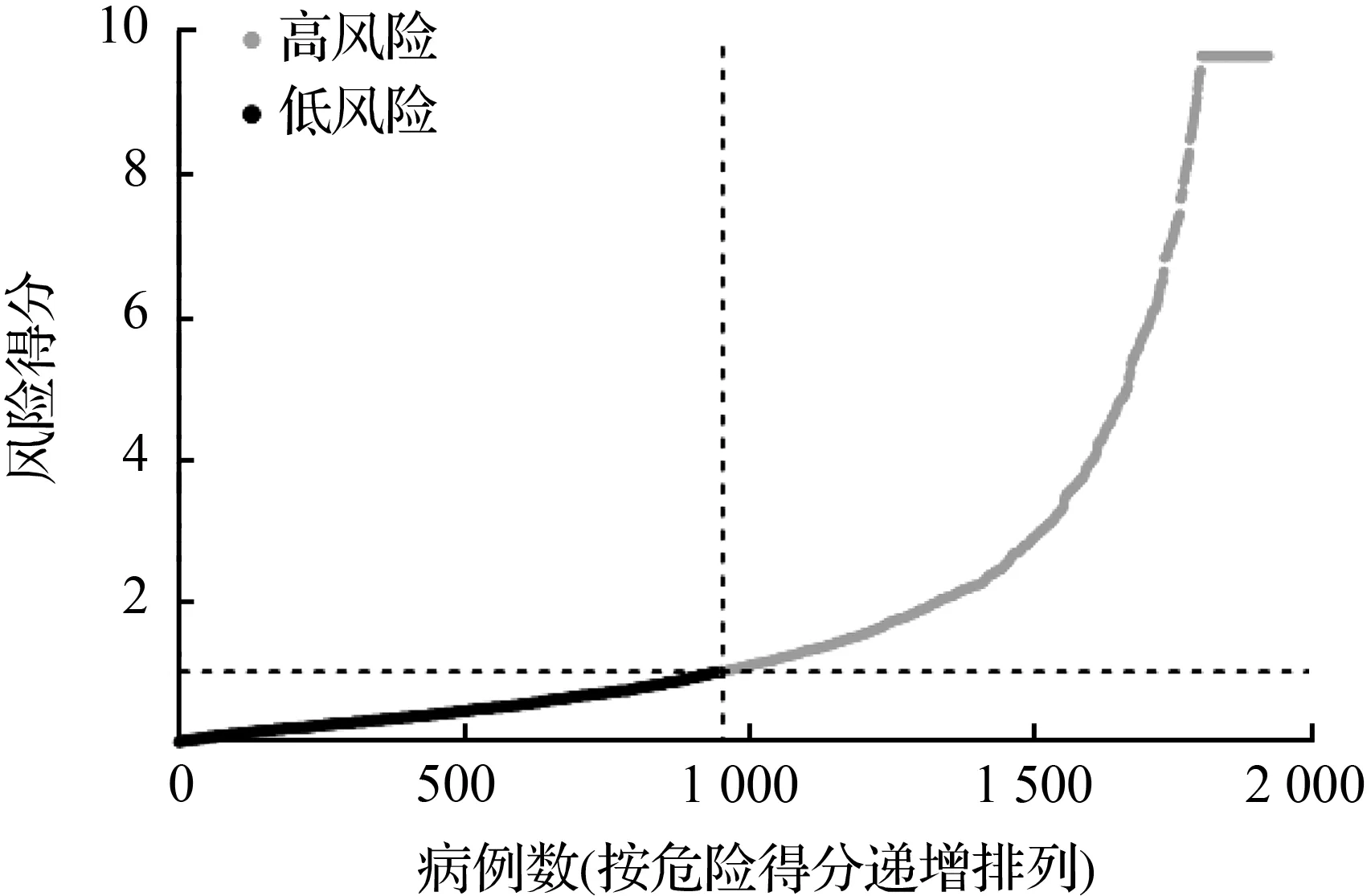
(a) METABRIC队列中风险评分的分布和中值

(b) METABRIC队列中OS状态、风险评分的分布

(c) Kaplan-Meier曲线, METABRIC队列中高风险组和低风险组患者的生存率

(d) ROC曲线下面积(AUC)评测预后模型
2.4 16个基因的独立预后分析
对可用得临床指标进行单因素和多因素Cox回归分析,以确定风险得分是否可以作为总体生存的独立预后预测因子。在单因素Cox回归分析中,年龄组(>=60 vs<60岁)显示HR=2.485,95 % CI=1.732~3.566,P<0.001; Stage组(Ⅲ/Ⅳ vs Ⅰ/Ⅱ)显示HR= 2.429,95 % CI=1.694~3.483,P<0.001;风险得分组(High vs Low)显示HR= 4.508,95 % CI=2.982~6.816,P<0.001,如图4(a)。在多因素Cox回归分析中,年龄组(>=60 vs<60岁)显示HR= 2.383,95 % CI=1.658~3.425,P<0.001; Stage组(Ⅲ/Ⅳ vs Ⅰ/Ⅱ)显示HR= 2.229,95 % CI=1.547~3.212,P<0.001;危险得分组(High vs Low)显示HR= 3.838,95 % CI=2.541~5.797,P<0.001,如图4(b)。由此可见,在校正了其他混杂因素后,年龄、Stage、危险得分均可以作为独立的预后因子。
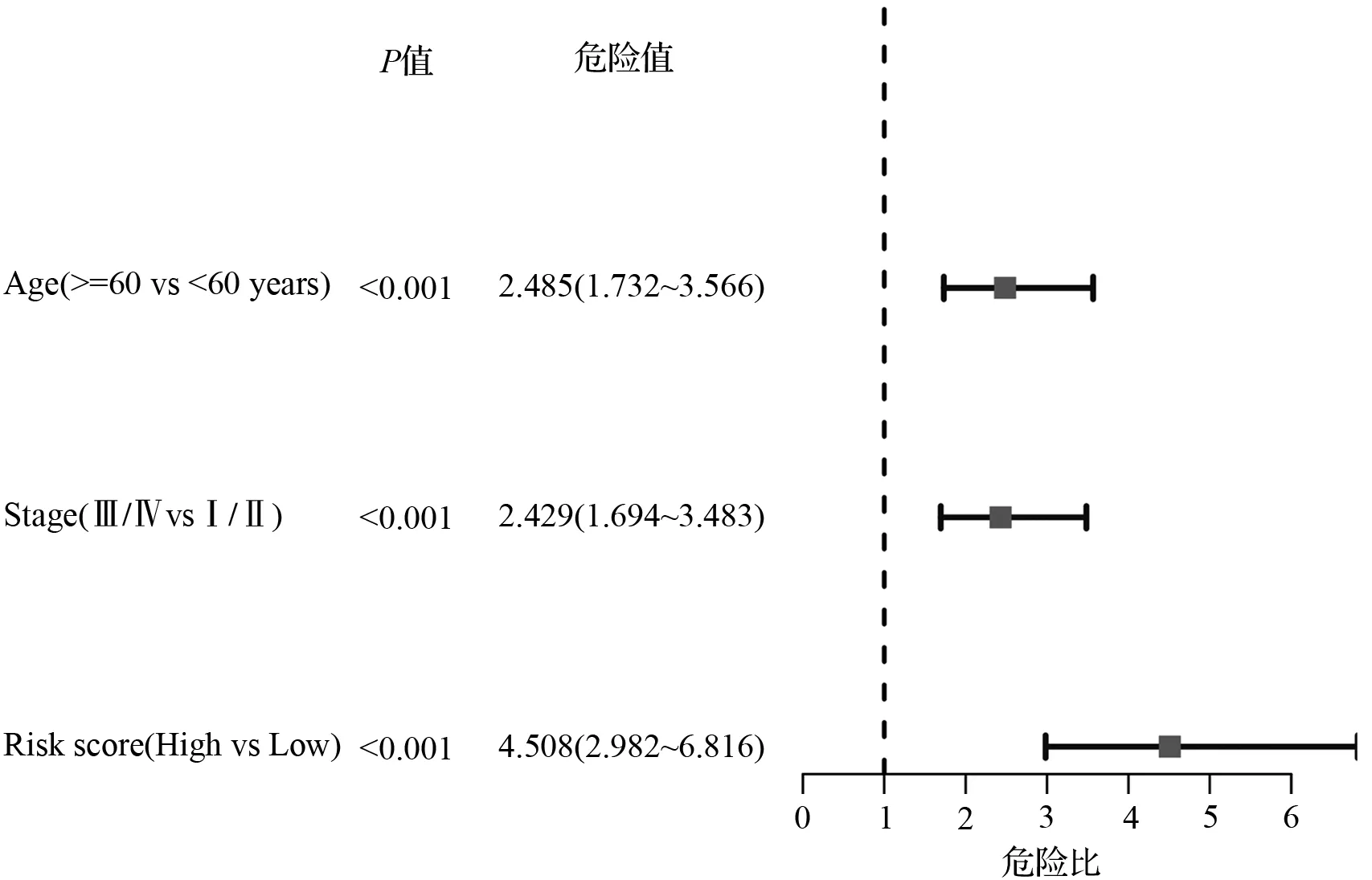
(a) 单因素回归分析
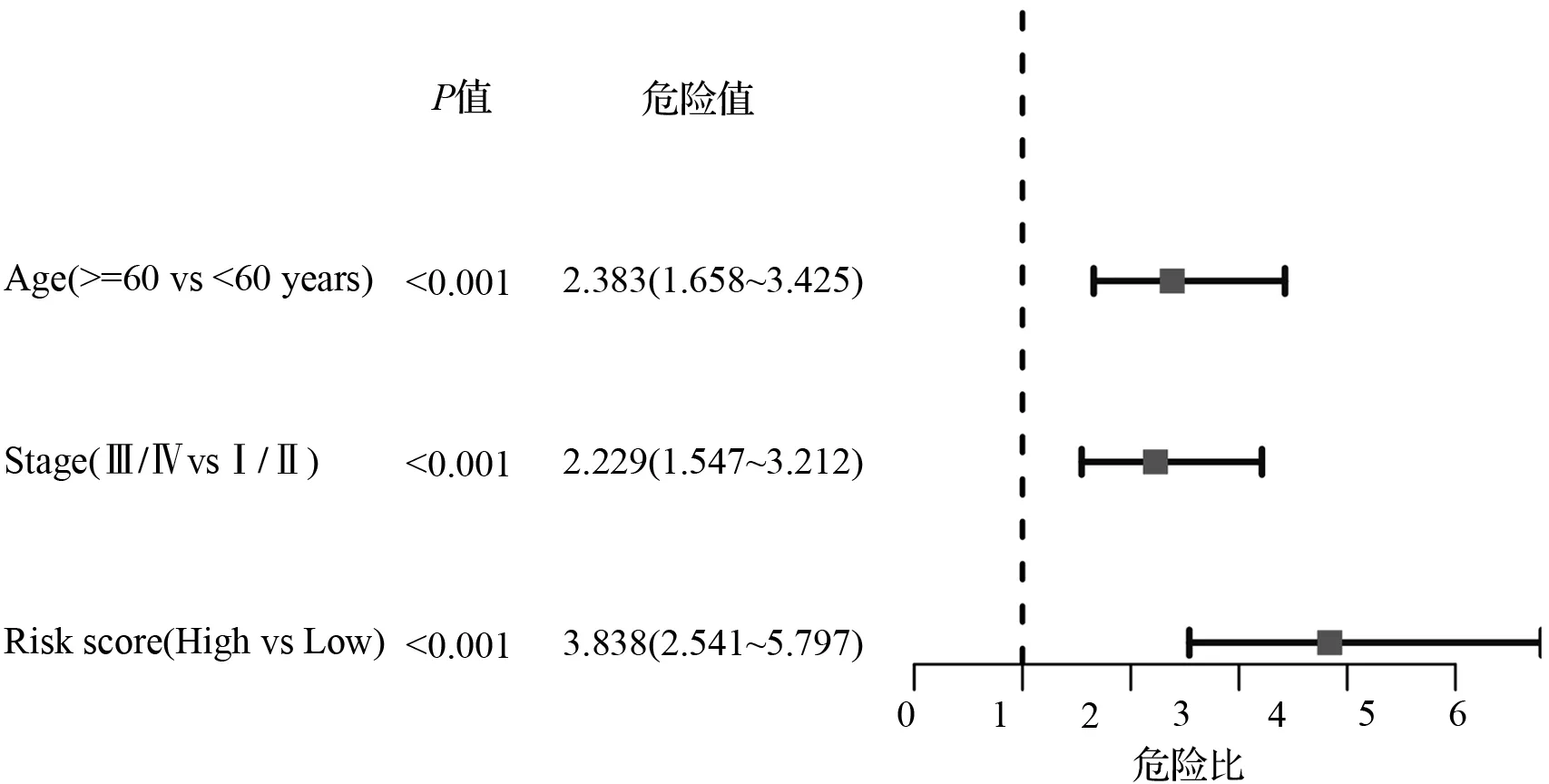
(b) 多因素回归分析
2.5 功能分析
为了探索与风险得分相关基因的生物学功能和机制,本研究采用高风险组和低风险组之间的差异基因进行GO富集分析和KEGG通路分析。在GO基因富集中,如图说是,差异基因富集在其中一个和铁相关的通路:金属离子的隔离。此外,还发现差异基因明显富集在了与免疫和炎症反应相关的通路,比如:抗菌体液反应,正向调节趋化性,白细胞趋化性,白细胞趋化的正调控,白细胞趋化的调节,调节趋化作用,并且所有的Padjust<0.05,如图5(a)。在KEGG功能富集分析中,如图5(b)所示,其中有3条和炎症和免疫反应机制相关的功能通路是明显富集到的,包括IL-17信号通路,PPAR信号通路,补体和凝血级联。

(b) KEGG分析
为了进一步探索在高、低风险两组间免疫相关的状态是否存在差异,通过ssGSEA分析对不同的免疫细胞和相关免疫通路进行免疫打分(图6)。如图6所示,在抗原呈递过程方面,发现aDCs、树突状细胞(DCs)、巨噬细胞(Macrophages)、APC_co_inhibition、APC_co_stimulation、MHC_class_I在高、低风险组之间存在显著差异,且高风险组的免疫得分高于低风险组。此外,ssGSEA分析中,炎症促进(Inflammation-promoting)和副炎症(Parainflammation)在高风险组的得分明显高于低风险组,这与KEGG富集在三条炎症相关的通路相对应。总之,在具有显著差异的结果中,除了Mast_cells和Type_II_IFN_Reponse在高风险组的评分低于低风险组,其他在高风险组的评分均高于低风险组。

(a) 16个免疫细胞
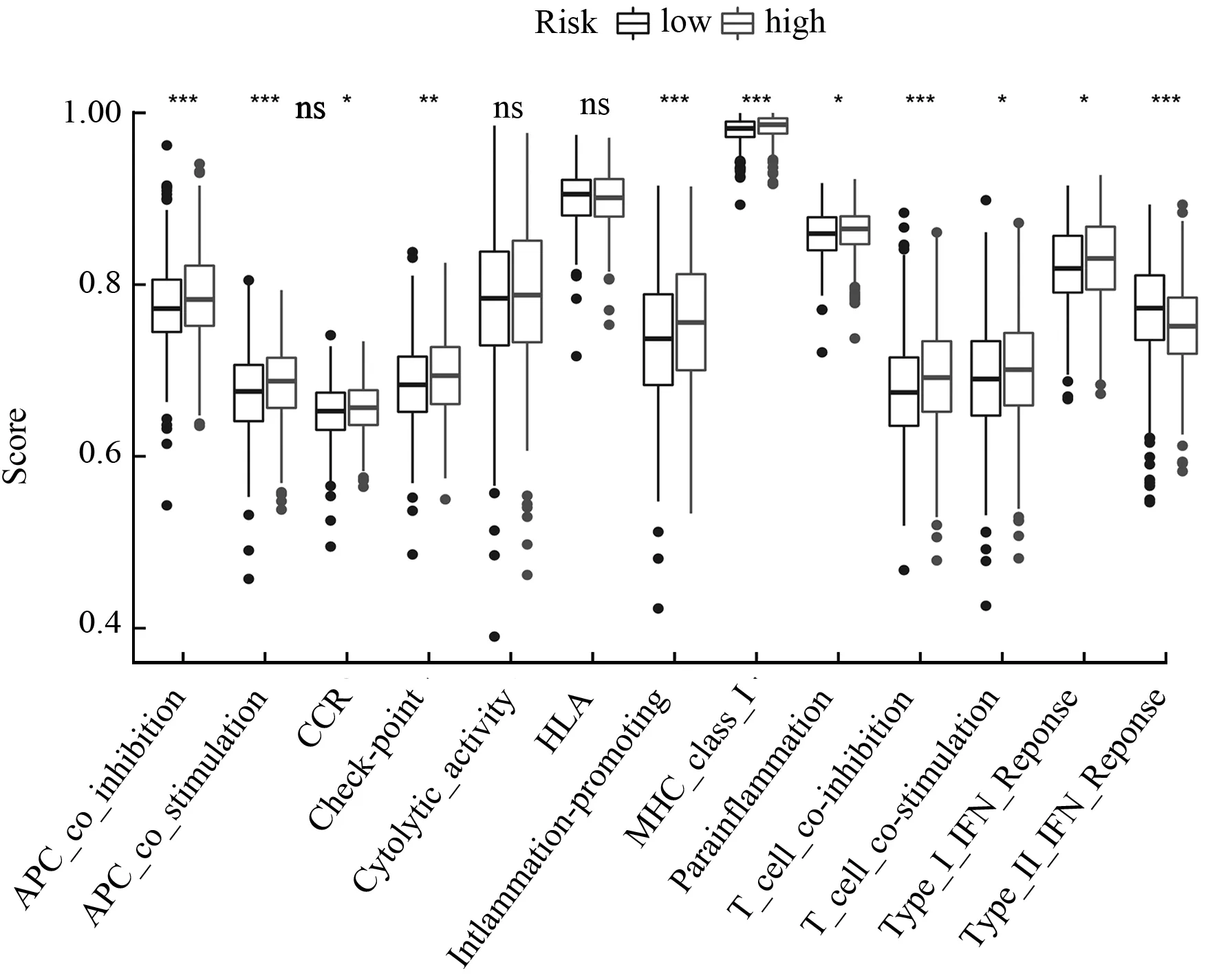
(b) 13个相关免疫功能
3 讨论
在本研究中,通过数据库挖掘和文献检索整合了148个和铁死亡相关的基因,并系统地研究了这148个铁死亡相关基因在乳腺癌肿瘤组织中的表达情况及其与生存预后的关系。关于16个铁死亡相关基因的预后模型首次被构建并在一个外部队列中被验证。此外,在功能分析中,发现了与免疫相关的通路被富集。
一些基因可能通过调节某种抑制剂来诱导或抑制肿瘤的铁死亡,但它们与患者生存预后的相关性仍不清楚。值得注意是,大约79.1 %的铁死亡相关的基因在乳腺肿瘤组织和癌旁组织之间有差异表达,且在这些差异基因中,有20.6 %的基因在单因素回归COX分析中具有显著差异,也就是说与生存预后相关。这表明了铁死亡在乳腺癌中具有潜在的作用,因此,通过铁死亡相关的基因建立乳腺癌预后模型是有意义的。
本研究构建的预后模型是由16个铁死亡相关的基因组成,包括CISD1、AIFM2、ALOX15、NCOA4、CHAC1、CS、GPX4、SQLE、CAPG、G6PD、NGB、GCLC、HSBP1、PRDX1、SLC1A4、BNIP3。
CISD1有调节线粒体中的铁和活性氧稳态的作用,通过调控mitoNEET蛋白维持线粒体稳态和促进肿瘤生长,对人类乳腺癌细胞增殖起着重要作用[12]。AIFM2在erastin等诱导癌细胞发生铁死亡的过程中起到了阻断作用[13]。ALOX15在c-MYC直接调控下,在肿瘤相关巨噬细胞中表达,其抑制作用可阻断原瘤基因的表达[14]。NCOA4负责将铁蛋白FTH招募到自噬小体从而使溶酶体降解铁蛋白、释放铁的运载蛋白从而可以抑制铁死亡[5],并且NCOA4又是乳腺癌的癌基因[15]。CHAC1通过 GCN2-eIF2α-ATF4 pathway通路利用降解谷胱甘肽来增强胱氨酸饥饿诱导三阴性乳腺癌细胞的坏死和铁死亡[16]。CS几乎存在于所有能进行氧化代谢的细胞中,在erastin诱导铁死亡中发生作用,主要是阻止其erastin诱导铁死亡[17]。由于GPX4具有将脂质过氧化氢转化为无毒脂质醇的作用,因此在很长一段时间内,GPX4一直被认为是抑制铁死亡作用的主要酶[18]。SQLE可能是乳腺癌的重要预后因子[19]。CAPG通过调节STC-1转录过程,增强乳腺癌转移发生率[20]。G6PD通过阻断内质网应激引起自噬并且G6PD通过抑制细胞自噬影响拉帕替尼对癌细胞的作用,此外与患者不良结局相关[21-22]。NGB的表达水平可作为 MCF-7乳腺癌细胞氧化应激的标志[23]。GCLC在功能上足以自主地产生他莫昔芬抗性代谢表型,比如:线粒体生物发生增加;ATP生产增加;谷胱甘肽水平降低。因此,药物抑制GCLC可能是解决乳腺癌患者对他莫昔芬产生耐药性的关键[24]。HSBP1在肿瘤组织中高表达影响了乳腺肿瘤的侵袭与迁徙,并且与患者的不良预后有关[25]。有研究表明,PRDX1下调可显著降低MCF-7和ZR-75-1乳腺癌细胞的生长速度[26]。雄激素受体信号通过增加谷氨酰胺转运体SLC1A4的表达促进谷氨酰胺代谢,这个基因在乳腺癌中经常过表达[27]。BNIP3在乳腺肿瘤发生中起抑癌作用,同时在某些乳腺癌的亚型中,也可作为肿瘤转移进展的预后指标[28]。
总之,这些基因在一定程度上对乳腺癌的进展有促进或者抑制的作用,但大部分基因是否通过铁死亡机制来影响乳腺癌患者的预后还尚不明确。
在以往的文献报道中,肿瘤对铁死亡易感性的机制一直是一个热门的研究领域,但肿瘤免疫与铁死亡之间的潜在调节机制仍不清楚。笔者对高、低风险间的差异基因进行了GO分析和KEGG分析,发现许多免疫相关的生物学通路和功能被富集。由此可推断,肿瘤免疫和铁死亡有密切的联系,或者说铁死亡很可能通过免疫机制影响了乳腺癌的进展。在ssGSEA分析中,发现抗原呈递过程在高、低风险组之间存在显著差异,这可能是铁死亡的细胞,通过释放一些信号,吸引抗原呈递细胞,从而发生抗原呈递过程激活免疫应激反应造成的[29]。此外,在高风险组中,Treg细胞、巨噬细胞以及免疫检查点的评分显著增高,这可能是Treg细胞在乳腺肿瘤组织中逐渐分化,浸润的Treg细胞的丰度越来越高,肿瘤恶化程度越来越深[30],而在恶性肿瘤中,既往研究已经证实,Treg细胞是其中重要的免疫抑制细胞[31];巨噬细胞的高评分可能是能够被吸附进入乳腺乳管中诱发一系列反应,将早期乳腺癌细胞离开乳腺组织扩散到机体其他部位,促进早期乳腺癌转移及进一步恶化的原因[32],还有研究发现巨噬细胞的新型亚群,称为表达平足蛋白的巨噬细胞(PoEMs),它可以改变肿瘤附近的组织,从而促进癌细胞的扩散[33];免疫检查点的高评分可能是乳腺肿瘤细胞能够利用被激活的免疫检查点通路来逃避免疫系统的识别,进而促进肿瘤的恶化[34]。由此可见,Treg细胞、巨噬细胞以及免疫检查点可能与乳腺癌的不良预后有关。
本研究还存在着一些局限性。首先,本文所建立的预后模型,受种族,地域,样本量等因素限制,因此,模型的稳健性还需进一步提高。其次,由于临床数据的限制,本文的研究没有涉及到乳腺癌的亚型研究,因此对于指导具体亚型的治疗策略的意义有待进一步研究。
总之,本研究通过构建16个与铁死亡相关基因预后模型,并确定了独立的预后因子,这为乳腺癌患者的治疗和预后提供了参考。
猜你喜欢
中老年保健(2022年6期)2022-08-19
核安全(2022年2期)2022-05-05
铁道建筑技术(2021年4期)2021-07-21
小学生学习指导(低年级)(2020年4期)2020-06-02
软件(2020年3期)2020-04-20
中国生殖健康(2019年2期)2019-08-23
中国生殖健康(2019年6期)2019-01-06
军营文化天地(2018年2期)2018-12-15
祝您健康(2018年5期)2018-05-16
产品可靠性报告(2017年7期)2017-09-05
