
气相色谱法测定婴幼儿配方乳粉中的OPO含量
2020-04-03 13:59张雪张丽茹王为蔡冬冬张筠
食品工业 2020年3期
张雪,张丽茹,王为,蔡冬冬,张筠*
1. 黑龙江东方学院食品与环境工程学部(哈尔滨 150066);2. 呼伦贝尔双娃乳业有限公司(呼伦贝尔 162750)
由于个人、社会等多方面外界因素的影响,许多地区出现了母乳喂养量缺乏、营养缺乏以及母乳喂养条件缺乏等情况,因此对母乳替代品——婴儿配方奶粉的研究备受重视[1]。婴儿配方奶粉中脂肪酸组成较牛乳更接近于人乳,但不能完全替代人乳,那么生产脂肪母乳化婴儿配方奶粉已成为乳品行业急待解决的重要问题[2]。1, 3-二油酸2-棕榈酸甘油三酯(简称OPO)现已经证实具有促进钙吸收和利用、促进婴儿发育、减少便秘和产梗阻发生的功能[3-6],所以近年来被广泛应用于保健食品、婴幼儿配方乳粉等领域。
中国人民共和国卫生部于2008年发布的第13号公告中,批准1, 3-二油酸2-棕榈酸甘油三酯(简称OPO)作为营养强化剂使用在婴儿配方食品、较大婴儿和幼儿配方食品中[7],并于2010年第1号公告中扩大其使用范围及使用量[8]。
目前,国内还没有建立关于检测婴幼儿配方乳粉中OPO含量的标准。国内外对于OPO的检测方法主要有气相色谱法[9-12]、液相色谱法[13-18]、气相色谱-质谱联用法[19-20]等,其中高效液相色谱法[21]是目前较广泛的方法,但使用的银离子色谱柱的稳定性差,柱子的使用率太低且寿命短,测定时间较其他方法长,回收率欠佳,并不适用于OPO的分离测定。结合实际情况,气相色谱法使用并不普遍,现有文献中的方法也并没有对方法进行深入研究,但气相色谱法较为稳定,测定的结果普遍准确且含量较高,因此试验拟建立一种测定乳粉中OPO含量的方法,旨在得到一种重现性好、精密度高且回收率高、结果准确的方法,使其越来越多地被应用到婴幼儿配方乳粉中OPO的定量检测中。
1 材料与方法
1.1 材料与试剂
25%氨水(分析纯,天津市致远化学试剂有限公司);无水乙醇(色谱纯,瑞典Oceanpak公司);无水乙醚(分析纯,天津市科密欧化学试剂有限公司);石油醚(优级纯,天津市科密欧化学试剂有限公司);正己烷(德国Merck公司);OPO标准物质(美国Sigma公司);十七烷酸甘油三酯标准物质(美国NU-CHEK公司);刚果红指示剂(浓度10 g/L);氮气;氢气(纯度≥99.995%);氧气;0.22 μm尼龙滤膜(有机相):上海安普生物科技有限公司;DB-1HT色谱柱(30 m×0.25 mm×0.1 μm,Agilent Technologies公司)。
1.2 仪器与设备
7890(FID)型气相色谱仪:Agilent Technologies公司;BT 125 D型电子天平(德国Sartorius公司);TST-UPB-20型超纯水机(泰斯特仪器有限公司);KQ 3200 DE型数控冲超声波清洗器(昆山市超声仪器有限公司);DKS-2型电热恒温水浴锅(泰斯特仪器有限公司);XW-80 A型旋涡混合器(其林贝尔仪器制造有限公司);RE-52 AA型旋转蒸发器(上海亚荣生化仪器厂);SPB-3全自动空气源(北京中惠普分析技术研究所);SPH-300 A氢气发生器(北京中惠普分析技术研究所)。
1.3 方法
1.3.1 标准溶液配制
OPO溶液(1 mg/mL):准确称取25 mg OPO标准物质,用正己烷定容至25 mL并使其充分溶解,在4 ℃条件下储存。
十七烷酸甘油三酯内标溶液(2.5 mg/mL):准确称取62.5 mg十七烷酸甘油三酯标准物质,用正己烷定容至25 mL并使其充分溶解,在4 ℃条件下储存。
混合标准工作液:准确移取100,200,300,400和500 μL OPO溶液,每份准确加入100 μL十七烷酸甘油三酯内标溶液,用正己烷定容至1 mL。其中OPO质量浓度为100,200,300,400和500 μg/mL,十七烷酸甘油三酯内标溶液质量浓度为250 μg/mL。
1.3.2 前处理条件优化
1.3.2.1 样品前处理步骤
试验选用已知OPO含量的奶粉进行测定,奶粉中的OPO含量是已知且稳定的,理论值为4.50±0.05 g/100 g。准确称取1.0 g(精确至0.000 1 g)样品,加入10 mL 65 ℃的超纯水,使其溶解,置于毛氏抽脂瓶中;加入35%的氨水,在一定温度下水解15 min;加入10 mL 95%无水乙醇以及2滴刚果红指示剂,再加入一定量的乙醚与石油醚;将合并后的有机相用无水硫酸钠脱水,在65 ℃下旋转蒸发至完全蒸干;将剩余的油脂用正己烷定容至25 mL,取2 mL上述液和1 mL内标溶液,用正己烷定容至10 mL,过0.22 μm滤膜,即为待测液。
1.3.2.2 碱水解液用量的选择
氨水溶液体系的加入可以破坏乳的胶体性以及脂肪球膜,使非脂成分溶解于氨水溶液体系中,而脂肪游离出来,参考GB 5009.6—2016第三法[22],选用1,2和3 mL的体积来判断氨水加入量对最终结果的影响。1.3.2.3 水解温度的选择
水解温度会直接影响碱水解液溶解脂肪的速度,但水解时间相差太短并不能对结果产生显著的影响,因此对水解温度进行优化,试验选择室温23 ℃与水浴65 ℃来判断水解温度对最终结果的影响。
1.3.2.4 萃取剂用量的选择
乙醚与石油醚作为萃取脂肪的萃取剂,其用量直接影响到脂肪的提取率。因此对萃取剂用量进行优化,选用以下两种方式来判断萃取剂用量对最终结果的影响:
1) 加入25 mL无水乙醚振摇1 min,静置分层后加入25 mL石油醚振摇1 min,静置分层后,将上层有机相转入鸡心瓶中,再加入5 mL 95%无水乙醇,加入15 mL无水乙醚振摇1 min,静置分层后加入15 mL石油醚振摇1 min,静置分层后,将上层有机相转入鸡心瓶中,最后加入15 mL无水乙醚振摇1 min,静置分层后加入15 mL石油醚振摇1 min,静置分层后,将上层有机相转入鸡心瓶中。
2) 加入25 mL无水乙醚振摇1 min,静置分层后加入25 mL石油醚振摇1 min,静置分层后,将上层有机相转入鸡心瓶中,再重复以上步骤提取2次。
1.3.2.5 萃取剂比例的选择
试验采用无水乙醚与石油醚作为萃取剂,萃取剂的比例同样影响萃取脂肪的结果,因此试验选用无水乙醚-石油醚比例分别为1︰1,1︰2和2︰1作为选择条件。
1.3.3 色谱条件优化
参照马小宁等[12]程序升温方法进行改进。分别采用两种程序升温方式进行色谱条件优化,进样样品为0.1 g/100 g添加内标的OPO标准品。升温程序(a):初始温度70 ℃,保持1 min,以50 ℃/min的速率升至170 ℃,再以20 ℃/min的速率升至350 ℃,保持25 min;升温程序(b):初始温度100 ℃,保持1 min,以50 ℃/min的速率升至300 ℃,保持1 min,以20 ℃/min的速率升至370 ℃,保持10 min。
进样口温度320 ℃;检测器温度370 ℃;载气为氮气,恒压模式;进样量1.0 μL;燃气流量30.00 mL/min。
1.3.4 OPO含量计算

式中:X表示试样中OPO的含量,g/100 g;C表示试样中OPO的浓度,mg/mL;V表示试样的定容体积,mL;m表示试样的质量,mg;f表示稀释倍数。
以重复性条件下获得的两次独立测定结果的算术平均值表示,结果保留两位有效数字。
1.3.5 方法验证
1.3.5.1 精密度
对同一添加OPO的乳粉样品,在相同的测定条件下进行6次重复试验,计算OPO含量以及相对标准偏差(RSD)。
1.3.5.2 加标回收率
称取一定量的样品,在3个加标水平下测定OPO含量,每个水平重复测定6次,计算回收率。3个水平的添加量分别为2.4,3.4和4.4 g。
1.3.5.3 标准曲线及相关性
移取100,200,300,400和500 μL OPO溶液,每份加入100 μL十七烷酸甘油三酯溶液,用正己烷定容至1 mL。配制成OPO质量浓度为100,200,300,400和500 μg/mL的系列溶液,对此混合标准溶液进行测定分析,以OPO含量为横坐标,峰面积为纵坐标作标准曲线。
1.3.5.4 检出限与定量限
称取一定量的样品,在相同的测定条件下进行3次重复试验,以相当于3倍信噪比待测液的量计算,得出OPO的最低检出限;以相当于10倍信噪比待测液的量计算,得出OPO的最低定量限。
2 结果与分析
2.1 前处理条件优化
2.1.1 氨水体积的选择
当加入1 mL氨水时,最终测得的OPO含量最高且最接近理论值,为4.36±0.02 g/100 g,最后选用1 mL氨水作为前处理最优条件之一,样品色谱分析图如图1所示。
2.1.2 水解温度的选择
在加入1 mL氨水的条件下,采用不同水解温度进行试验。由计算可知,当在65 ℃温度下水解时,最终测得的OPO含量最高且最接近理论值,为4.45±0.03 g/100 g,最后选用在65 ℃温度下水解作为前处理最优条件之一,样品色谱分析图如图2所示。

图1 加入1 mL氨水样品色谱图

图2 65 ℃水解样品色谱图
2.1.3 萃取剂用量的选择
在加入1 mL氨水、65 ℃温度水解的条件下,采用不同萃取剂用量进行试验。由计算可知,当采用萃取剂用量(a)时,最终测得的OPO含量是最高的,为4.46±0.03 g/100 g,同时在试验过程中发现,采用萃取剂用量(b)时,萃取过程中会产生大量白色浑浊物体,沉淀不完全,导致肉眼无法观察萃取情况,影响后续试验操作与上机分析。综上所述,最后选用萃取剂用量(a)作为前处理最优条件之一,样品色谱分析图如图3所示。

图3 萃取剂用量(a)样品色谱图
2.1.4 萃取剂比例的选择
试验以不添加OPO原料的婴幼儿配方乳粉作为基质粉,准确称取1 g基质粉,向其中加入3 g OPO标准品,以无水乙醚与石油醚的比例分别为1︰1,1︰2和2︰1为变量条件测定OPO含量,每个变量重复测定6次,测得结果如表1所示。

表1 以萃取剂比例为变量试验结果 g/100 g
基质粉中OPO含量如图4所示。已知基质粉中OPO含量约为1.72 g/100 g,加入标准品后的奶粉OPO理论含量约为4.72 g/100 g。表1表明,当无水乙醚︰石油醚比例为1︰1时,测定结果最接近于最终理论值。综合比较,选择无水乙醚与石油醚的比例1︰1作为前处理最优条件之一。
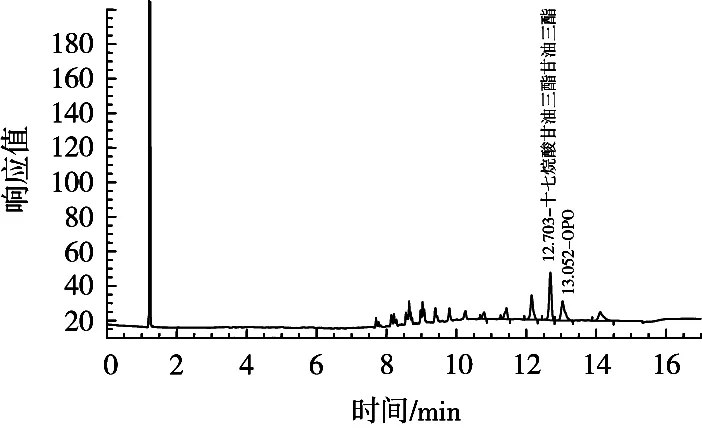
图4 基质粉OPO色谱图
综上所述,前处理条件的最终选择为:氨水体积1 mL、水解温度65 ℃、萃取剂用量(a)、无水乙醚与石油醚的比例1︰1。
2.2 色谱条件优化
程序升温(a)色谱图如图5所示。采用(a)程序升温,内标物峰与目标峰的出峰时间均在13 min左右,内标物峰与目标峰分离效果不理想且峰型不好,杂峰较多,无法准确定位内标物峰与目标峰,为定量分析带来困难。程序升温(b)色谱图如图6所示。采用(b)程序升温,内标物峰与目标峰的出峰时间均在12 min左右,内标物峰与目标峰分离效果理想且峰型良好,几乎无杂峰,可以准确定位内标物峰与目标峰,适用于测定婴幼儿配方乳粉中的OPO含量。
2.3 精密度测定
对同一添加OPO的乳粉样品进行6次重复试验,计算OPO含量并进行精密度分析,计算其相对标准偏差(RSD),结果见表2。RSD值为2.57%,小于5%,表明仪器及方法的精密度与重现性良好,测定婴幼儿配方乳粉中的OPO具有较高的准确度。
2.4 加标回收率测定
准确称取0.1 g样品,依次添加3个不同水平的OPO标准品,每个水平重复测定6次,按上述试验条件操作,计算回收率,结果如表3所示。结果表明,加标回收率平均为92.97%±5%,可以满足分析方法对于加标回收率的要求,可以证明采用该方法测定婴幼儿配方乳粉中的OPO具有较高的准确度,效果良好。
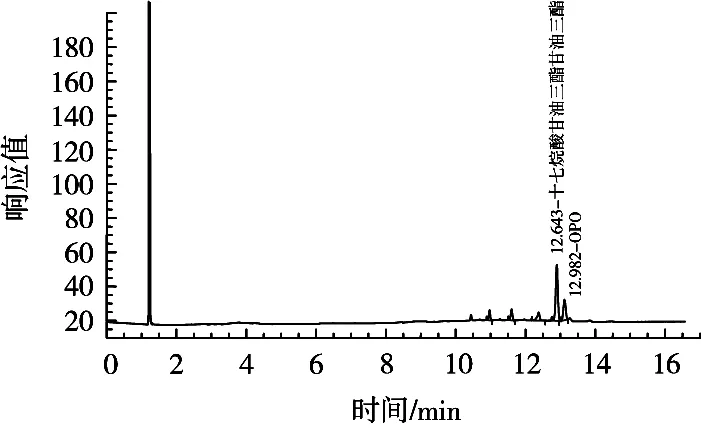
图5 采用程序升温(a)色谱图
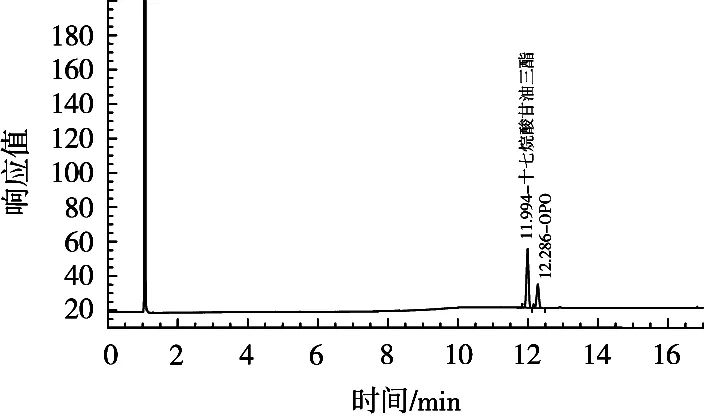
图6 采用程序升温(b)色谱图
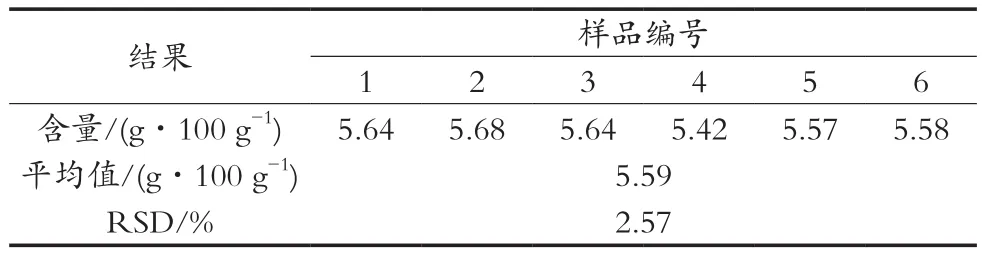
表2 精密度测定结果

表3 回收率测定结果
2.5 标准曲线及相关性
对不同浓度的OPO混合标准溶液进样,进行线性回归,结果如图7所示。可以看出标准曲线浓度在0.1~0.5 g/100 g范围内,含量与峰面积线性关系最好,证明该方法响应值与校正曲线一致度高,线性回归方程为y=583.34x-13.858,相关系数R2=0.999 8。
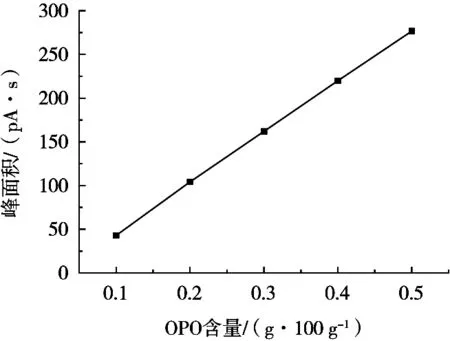
图7 OPO标准曲线
2.6 检出限与定量限测定
以相当于3倍信噪比待测液的量计算,得出OPO的最低检出限为0.04 g/100 g;以相当于10倍信噪比待测液的量计算,得出OPO的最低定量限为0.14 g/100 g。
3 结论
试验采用气相色谱法测定婴幼儿配方乳粉中OPO的含量,通过乙醚与石油醚提取乳粉中的脂肪,经过无水硫酸钠脱水、旋转蒸发浓缩等处理,结果表明其RSD为2.57%,回收率为92.97%±5%;在0.1~0.5 g/100 g范围内,R2为0.999 8,检出限为0.04 g/100 g,定量限为0.14 g/100 g。该方法操作简便、精密度高、回收率良好、结果准确,适用于婴幼儿配方乳粉中OPO含量的检测。
猜你喜欢
食品科学(2023年4期)2023-03-06
食品安全导刊(2021年21期)2021-08-30
商情(2020年47期)2020-12-15
中国乳业(2020年12期)2020-04-12
佳木斯大学学报(自然科学版)(2020年1期)2020-02-28
科技视界(2019年12期)2019-06-20
百家讲坛(2019年24期)2019-04-24
农业与技术(2018年20期)2018-11-16
中成药(2017年12期)2018-01-19
农产品加工(2017年6期)2017-05-09
