
异烟肼片仿制药质量研究和参比制剂的选择*
2020-03-25 00:55刘小燕张晓明张义福杨平荣
甘肃科技 2020年24期
刘小燕,王 娟,张晓明,常 琦,张义福,杨平荣
(1.甘肃省药品检验研究院,甘肃 兰州 730070;2.甘肃中医药大学,甘肃 兰州 730000)
仿制药质量的差异直接影响群众用药的安全性和有效性。为整体提升我国药品质量水平,《国家药品安全“十二五”规划》明确提出把全面提高仿制药质量作为一项重要任务[1]。异烟肼,化学名4-吡啶甲酰肼,是结核病联合治疗中的首选药。本品分子量小137.14,口服后几乎完全吸收,生物利用度达90%[2]。国内异烟肼片批准文号数571 个,涉及生产企业331 个,有500mg,300mg,100mg,50mg 四个规格,临床首选规格为100mg。属于289 目录品种之一[3]。截至3 月13 日,异烟肼片参比制剂备案记录共52条,涉及生产企业数24 个。有12 家企业申报一致性评价,通过一致性评价的有6 家企业,涉及8 个受理号,包括100mg6 个,300mg2 个。目前,均已纳入《中国上市药品目录集》。笔者以异烟肼片为例,对异烟肼片仿制药体外质量研究进行了总结分析,并对参比制剂的选择提出建议,期望对我国仿制药质量一致性评价提供参考。
1 品种概况
根据美国儿童健康和人类发展研究所(NICHD) 和美国FDA BCS(2011 年)分类[4]及WHO(2005 年)的研究[5],异烟肼为BCSⅠ或BCSⅢ类。报道指出异烟肼不存在多种晶型[6]。FDA 和WHO 指出,经过长期的临床有效性验证,异烟肼片可以通过体外溶出曲线的方法来评价与参比制剂的一致性,不推荐体内试验,可以生物等效性(bioequivalence,BE)豁免[6,7]。国家药品监督管理局(NMPA)也提出异烟肼片可申请豁免人体BE 研究[8]。因此,体外药学研究则是评价异烟肼片一致性最主要的手段。
NMPA 发布的异烟肼片100mg,300mg 推荐的参比制剂为FDA 指定的参比制剂(RLD):SANDOZ INC(山德士),现已撤市。针对这种RLD 不能获得的情况,FDA 于2017 年引入了对照标准制剂(RS)。其中异烟肼片的RS 为BARR LABORATORIES INC。FDA 指定RS 仅用于作为仿制药申请(abbreviated newdrugapplication,ANDA)时进行BE 研究的对照制剂,体外药学研究还应与RLD 进行对比[9]。NMPA 之后又增补了 BARR LABORATORIES INC/Teva Pharmaceuticals USA,Inc.(梯瓦)异烟肼片同时作为我国参比制剂之一。目前50mg,500mg 异烟肼片没有推荐参比制剂。
2 仿制药质量研究
2.1 杂质分析
异烟肼合成路线主要有两条,如图1 所示[10]。
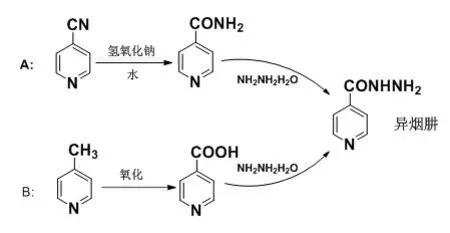
图1 异烟肼合成路线图
异烟肼及其片剂主要已知杂质有8 种,见表1。除了未反应的起始原料,在合成过程中会进一步产生的反应中间体,降解产物,副产物及相容性产物等。

表1 异烟肼及其片剂已知杂质及归属
2.1.1 游离肼
游离肼可在原料合成时引入,也可在储存过程中由异烟肼降解产生,肼是一种诱变剂和致癌物质[11]。因此,对游离肼含量的控制是评价异烟肼及其制剂质量的重要指标之一。国内外标准中有三种检测方法:浊度法、TLC 及柱前衍生HPLC 法,见表2。

表2 异烟肼及异烟肼片国内外标准比较
仅ChP2015 在异烟肼及其片剂中都设有游离肼检查项。其中JP17 目视浊度法不能客观反映游离肼真实含量,而ChP2015 采用的TLC 法检测灵敏度较低。此前,中国国家药典委员会已拟定提高异烟肼及其制剂质量标准[12]。
HPLC 法是利用游离肼与苯甲醛反应生成的苯甲醛吖嗪进行色谱分析测定,该方法准确性好,专属性强,灵敏度高[13]。已有国内企业在申报异烟肼片一致性评价的注册标准中将游离肼测定法修订为HPLC 法,设定限度低至0.0015%,等同于BP2020/EP10.0 对原料药中游离肼的限度:15ppm。这或许预示着我国仿制药质量在杂质控制方面有了较大提高。客观、合理的质量控制限度的制定,还需结合仿制药与参比制剂稳定性试验中游离肼的测定数据,以保证产品在有效期内达到限度要求。
2.1.2 有关物质
1)杂质谱分析。有关物质是药品关键质量属性之一,对药品安全性和质量可控性有着重要影响。国内外药典对异烟肼有关物质的质量控制差异较大。比较之下,BP2020/EP10.0 的质控限度较为严格。仅ChP2015 在异烟肼片中设定有关物质检查项,其余药典均未在片剂中控制有关物质(见表2)。
在前期研究工作中,笔者测定了包括异烟肼和山德士、日本第一三共在内的20 家企业异烟肼片的有关物质。山德士异烟肼片检出一特有杂质,含量高达3.1%,经LC-MS 结构确证为异烟肼与乳糖原辅料相容性产物——乳糖异烟腙。而17 批国内异烟肼片检出未知杂质在0.2%以上的有2 家,涉及3 种杂质;异烟肼及所有异烟肼片中均检出异烟酸、异烟酰胺,而4-甲基吡啶及USP43 与BP2020/EP10.0 列出的4-氰基吡啶,2-吡啶甲酰肼均未检出[14]。基于以上研究,并结合文献资料对8 种已知杂质进行了来源归属,详见表1。一般情况下,工艺杂质及降解杂质都须在原料中进行控制,由于制剂中工艺杂质不会增加,故只须控制降解杂质及原辅料相容性产物。在异烟肼片仿制药一致性评价中,已知杂质中须控制的是:异烟酸、异烟酰胺、1,2-二异烟酰基肼及乳糖异烟腙。如果研究证明光降解产物1,2-二异烟酰基肼在确定的储存条件下,并在加速试验、长期试验中不会增加,则在质量标准中无需单独控制,笼统要求即可。乳糖异烟腙仅在处方中添加乳糖时产生,没有添加时无需考虑。
异烟肼片最大服用量为900mg/天,依照ICHQ3b 中对于制剂杂质的要求,杂质报告限为0.1%,鉴定限为0.2%,质控限为0.2%[15]。企业需对检出在0.2%以上的未知杂质进行结构确证,参考文献资料或根据毒理实验结果确定质控限。但对仿制药研发而言,企业可以从提升原料质量,优化制剂生产工艺角度入手,降低该杂质含量至鉴定限以下。
2)限度的制定。对于已知杂质,如异烟酸、异烟酰胺及乳糖异烟腙,有质量标准或文献资料的,可结合参比制剂与仿制药稳定性试验结果确定杂质限度。杂质对照品价格昂贵,不建议外标法测定,主张采用加校正因子的主成分自身对照HPLC 法进行定量。为确保相对保留时间定性的准确性,质量标准中可指明色谱柱型号及参数。在系统性试验中,需要验证杂质与主成分之间的分离度时,可采用杂质对照品的相应试剂代替。未知杂质的控制,研究者可根据稳定性试验数据统计分析,以帮助拟定质控限度,试验设计参照原CFDA 发布的《化学药物(原料药和制剂)稳定性研究技术指导原则》。
如果该杂质为仿制药特有杂质,质控限就不得高于鉴定限,对异烟肼片来说是0.2%。若为仿制药与参比制剂共有杂质,当参比制剂含量高于鉴定限0.2%时,质控限不得高于参比制剂含量;若参比制剂含量低于鉴定限0.2%,则仿制药可高于参比制剂,但不得高于鉴定限。为使降解杂质符合质量标准限度,缩短有效期是被容许的,国家药品审评中心(CDE)公布的已通过异烟肼片一致性评价的产品说明书中有效期存在12 个月,24 个月。而一致性评价之前,多数国内企业异烟肼片有效期设定均为36个月。杂质质控详细研究思路可参考谢沐风作者的系列文章[16,17]。
2.2 溶出度
体外溶出行为的比较已成为仿制药申报和上市后质量监管工作中越来越重要的一个关键质量指标。异烟肼片为可申请BE 豁免品种,需证明仿制制剂与参比制剂为BCSⅠ类快速溶出或BCSⅢ类非常快速溶出。此时,胃排空是药物吸收的限速步骤,药物的吸收速度和吸收程度就不会依赖于药物的溶出时间或在胃肠道的通过时间[18],仅取决于制剂中API 的特性[19,20]。李飞[21]认为对于高溶解性快速溶出类制剂的处方、原辅料质量及生产工艺的变化很难从溶出结果中体现,此类产品溶出方法的确定不需要更多验证。
然而,申请生物豁免并非易事。BCSⅢ类药物的难点在于达到仿制制剂与参比制剂处方种类用量的一致;BCSⅠ类药物的难点在于证明其高渗透性。而渗透性数据的获得并不容易,有些渗透性试验的成本甚至超过了体内BE 试验[22]。所以我们才会看到,在申报异烟肼片一致性评价时,有两家企业放弃申请生物豁免,完成了BE 研究。
一般来说,选择原研制剂的溶出度方法是一种比较保险的做法[23]。但是有的情况下,选择已有溶出方法作为质量标准并不能很好反映产品的关键质量属性[24]。依据“仿产品,不是仿标准”理念,还应根据仿制制剂的特点及BE 研究数据,在对参比制剂、仿制药进行溶出曲线研究后,制定出符合本制剂质量控制的溶出度方法作为该仿制药上市后的质量标准[25]。
笔者之前的pH-溶解度曲线测定显示异烟肼的溶解度无pH 依赖性[26],该曲线与X 轴基本平行,理论上同一制剂多条溶出曲线应重合。这与我们对山德士异烟肼片及国内多家企业仿制制剂在四种溶出介质中的溶出曲线测定结果一致[27]。同时,我们可以假设,若仿制制剂本身四条曲线不能重合,推测产品的处方工艺还有提升空间。在测定了20 家国内异烟肼片的溶出曲线后,结果显示不同企业制剂在水中溶出量差异较大[26]。比较各国药典除USP43 溶出介质为0.01mol/L 盐酸溶液外,其余均为水。日本的“药品品质再评价工程”将溶出度评价作为主要手段,提出“溶出一致,临床疗效一致”理念,溶出试验条件也是各国药典中最严格的[28,29]。我们有理由相信JP17 的溶出度质量标准具有更强的可参考性。溶出装置选择桨法,转速为50rpm。同时,从秉承环保、提高效率出发,首选水作为溶出介质。
溶出曲线测定时,为保证不同采样时间点溶出量测定的准确度,通常采用HPLC 法;而在质量标准拟定时,快速溶出类制剂通常为一个介质一个时间点,在排除辅料干扰的前提下,考虑UV 对照品法。溶出限度的制定可参考FDA 发布的相关指南[30],在规定的溶出条件下,API 为高溶解性的口服固体制剂的可接受限度为30min 溶出量80%,以此作为一个最低的要求。
3 参比制剂的选择
参比制剂是一致性评价的标杆,对体外质量评价有决定作用。参比制剂应是处方工艺合理,质量稳定的药品。
3.1 处方组成
参考药品说明书,山德士与梯瓦异烟肼片(100mg)添加辅料的功能基本一致,最大的不同是山德士异烟肼片处方中添加了辅料乳糖(见表3)。

表3 参比制剂辅料比较
乳糖是片剂较理想的稀释剂,我国乳糖产量低,价格贵,较少使用。调研发现我国仿制药异烟肼片处方中均不含乳糖。而文献报道,乳糖可与胺类化合物配伍后生成棕黄色的加成物,在处方中含有乳糖时,可能会增加异烟肼片的生物豁免风险[31,32]。
3.2 质量分析
在杂质谱分析中,发现山德士异烟肼片中的特有杂质乳糖异烟腙,含量可高达3.1%。该杂质含量远远超出了ICH 中Q3b 的质控限度[15]。同时,山德士异烟肼片有关物质两批批间差异较大,批号ME130402:异烟酸0.21%,乳糖异烟腙1.6%,异烟酰胺0.12%,各杂质总和2.0%;批号ME130036:异烟酸0.06%,乳糖异烟腙3.1%,异烟酰胺0.03%,各杂质总和3.2%[14]。
笔者采用已建立的HPLC 溶出量测定方法[27]:桨法50rpm,溶出介质为pH1.2,pH4.5,pH6.8 及水500ml,取样时间点为5min,10min,15min,30min。测定了两种参比制剂在四种溶出介质中的溶出曲线。结果显示,山德士与梯瓦异烟肼片在四种介质中15min 时溶出量均达到85%以上,为非常快速溶出(如图2 所示)。
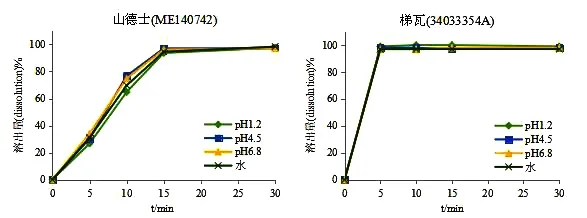
图2 参比制剂在四种溶出介质中的溶出曲线
同一批12 片样品15min 溶出量RSD%,梯瓦:RSD%<2%(n=12),山德士:RSD%<4%(n=12)。梯瓦异烟肼片批内一致性更佳。
3.3 小结
从参比制剂遴选顺序来说,原研产品具有全套的质量研究、体内BE 试验及临床有效性试验数据,应首选FDA 进口原研制剂:山德士异烟肼片。但作为可申请生物豁免品种,在符合快速溶出或非常快速溶出时,生物不等效风险较低。在经过一系列药学研究后,显示山德士处方存在异烟肼与乳糖不相容的风险,有关物质中检出含量3.1%的杂质;而梯瓦异烟肼片的处方合理,溶出均一性更佳,在结合稳定性试验数据的基础上,可优先考虑作为我国异烟肼片一致性评价参比制剂。
4 结语
仿制药一致性评价研究的核心是对药物制剂处方工艺的再研究。不论是申报仿制药还是一致性评价,目标都是建立一个完善合理的质量保证体系[24],保障仿制药上市前后产品质量与参比制剂或原研制剂一致。NMPA 第102 号文指出,自首家仿制药品种通过一致性评价后,其他生产企业的相同产品原则上3 年内要完成,在药品集中采购方面不再选用未通过一致性评价的品种[33]。同时,“4+7 带量采购”的实施,使通过一致性评价的仿制药与原研产品同组竞价[34,35],促使仿制药企业完善生产线,实现行业全面转型升级。
近年抗结核菌药物导致的严重肝损伤越来越受到人们的关注,异烟肼作为一线药物有着不可替代的作用。在新一代抗结核药物问世前,如何利用现代化技术,研发出能减少异烟肼造成肝损害的新型药物制剂显得越来越迫切[36]。中国已加入ICH,对国内药企的研发能力将提出更高的要求,对企业来说既是机遇也是挑战。企业在积极应对的同时,应全面做好战略布局,应对产业升级,不断研发创新。
猜你喜欢
化工时刊(2022年4期)2023-01-06
江苏卫生保健(2022年5期)2022-05-24
山东化工(2020年20期)2020-11-25
幸福(2018年33期)2018-12-05
中成药(2018年9期)2018-10-09
中成药(2018年7期)2018-08-04
中成药(2018年7期)2018-08-04
中外医疗(2015年11期)2016-01-04
饮食科学(2015年4期)2015-11-28
中国卫生标准管理(2015年13期)2015-01-26
