
第一性原理计算Fe掺杂LiMgP新型稀磁半导体的半金属铁磁性
2020-03-17 06:30杜颖妍向朝凯杜成旭毋志民
功能材料 2020年2期
陈 婷,庞 军,何 红,杜颖妍,向朝凯,贾 倩,刘 焦,于 越,杜成旭,毋志民
(重庆师范大学 物理与电子工程学院,光电功能材料重庆市重点实验室,重庆 401331)
0 引 言
在现代信息技术中,对信息的处理和存储分别利用的是半导体电子的电荷自由度和磁性材料的自旋自由度[1]。若能将电子的电荷特性和自旋特性集于一体,将其应用于信息技术中,将有望开发出集成度更高、运行速率更快、能耗更低的新型电子器件。而自旋电子学(Spintronics) 正是操作半导体中的电子自旋和电荷两个自由度来进行对信息的加工和处理[2]。半金属性铁磁体 (half-metallic ferromagnet)是一个自旋方向的电子能带具有金属性而另一个自旋方向的电子能带具有非金属性的磁性材料。由于电子结构的这一特性使它们在费米面处的自旋极化率(spin polarization)为100%,被认为是制作下一代电子器件——自旋电子学器件的理想材料[3-5]。在半金属材料中Heusler合金因为其具有较高的居里温度(TC)、较大的磁矩、较宽的半金属能隙[6]和可控的电磁性质而被前人广泛的研究。
而稀磁半导体恰好能够同时利用半导体中电子的电荷和自旋属性,是新一代自旋电子学器件的关键材料,但是在II-VI族半导体中,Mn离子的不等价替换只引入了局域磁矩,不同的磁性原子浓度和不同温度下的磁性行为各异,且掺杂成n型或p型是非常困难的[1]。传统的III-V族稀磁半导体的磁性离子不等价替代只能形成亚稳态薄膜材料,严重限制了材料的固溶度[7],且制备方法直接影响着材料的质量[8]。而Mašek等[9]和Jin等[10]通过理论和实验发现了一类与半Husler合金同为类闪锌矿结构的I-II-V族新型稀磁半导体Li1±y(Zn1-xMnx)As,它可以实现自旋和电荷注入机制的分离调控,由Mn2+替换Zn2+来注入自旋,而改变体系中Li的含量则能够调控体系中的载流子浓度。Li(Zn,Mn)As虽然有许多优异的性能,但遗憾的是TC有50 K,远小于300 K的要求。Sato等人[11]研究了Mn掺LiZnX(X= As, P, N)体系的电子结构,结果发现Li空位不仅能够提高体系的稳定性和TC,还能诱导Mn离子间的铁磁交换作用.Deng等[11]发现Li(Zn,Mn)P为软磁性材料,具有较低的矫顽力,增加体系载流子浓度能够提高TC,由电荷和自旋分离掺杂的方法能够合成该体系。Kacimi等人[12]采用第一性原理计算方法发现LiMgP较其他半导体具有更宽的带隙,有望开发出性能优异的稀磁半导体材料;Sato[13]等人、Venkatesan[14]小组、Lin[15]等人和Potzger[16]等人的研究表明Fe掺杂的ZnO具有稳定的铁磁性。因此,本文选取LiMgP为基体,采用第一性原理广义梯度近似计算方法[17],系统的研究了不同Li含量下Fe掺杂LiMgP体系的能带结构、电子态密度、差分电荷密度和轨道电子数,结果表明Fe单掺具有大的半金属能隙,是一种性能优异的半金属铁磁体,且调节体系Li含量能够调节载流子浓度,这为Li(Mg,Fe)P在自旋电子学器件方面的应用提供了理论依据。
1 计算模型与方法
1.1 计算模型
计算选用的理想模型是LiMgP反萤石结构[18],属于F-43m空间群,晶格常数a=b=c=0.6005nm,其中c/a=1.000。LiMgP可由Li,Mg和P反应生长后在高温(830℃)下结晶析出[18]。根据“间隙插入规则”[20],LiMgP可看作是一个在邻近P四面体间隙填上Li的二元复合[MgP]-闪锌矿结构。LiMgP是直接宽禁带半导体,带隙值为2.43eV[18]。研究采用24原子(2×1×1)超胞模型,如图1所示,掺杂时用Fe原子取代LiMgP中最高对称位置上的Mg原子。为调节体系中的载流子浓度和寻求最稳定结构,分别构建了Li过量和Li不足时,3种不同Li空间占位的超晶胞,对应的结构如图1(c) 和(d)的ILi1、ILi2、ILi3和VLi1、VLi2、VLi3所示。

图1 Li1±y(Mg1-xFex)P的超晶胞结构
1.2 计算方法
本文采用第一性原理的方法,在MS的CASTEP[21]量子力学模块下完成相关计算。电子波函数采用平面波超软赝势法[23],利用广义梯度近似(GGA)中的PBE[22]修正.体系能量和电荷密度的积分计算采用 Monkhorst-Park[24]方案,K-point设置为5×5×5,平面波截断能(Ecut)为520eV,体系中Li,Mg,P,Fe 各原子参与计算的价电子分别为Li:2s1,Mg:2p63s2,P:3s23p3,Fe:3d64s2,其自洽收敛精度设为2.0×10-6eV/atom。晶胞结构优化后,各项参数均优于收敛标准。对LiMgP基体,采用GGA优化后的带隙值与实验值更接近、总能更低,所以采用GGA的计算方法更合理[18]。掺杂体系的稳定性可根据形成能Ef来判断[25]:
Ef=E[Li1±y(Mg1-xFex)P]-E[LiMgP]-nFeμFe+
nMgμMg±nLiμLi
(1)
其中E表示计算的体系总能,n为改变的原子个数,μ为化学势,化学势的取值本与实验条件相关,这里旨在比较各体系基态的相对稳定性,所以仅在电中性情况下选取Fe,Mg,Li的化学势为各自稳定结构下的单质能量。通过分析优化后的晶格常数、形成能和总能发现(如表1所示)掺杂体系的稳定性均有所提高,且ILi2和VLi2为最稳定位置,最后选取这两种结构计算不同Li含量体系的性质。

表1 Li1±y(Mg1-xFex)P的晶格常数、总能和形成能
2 Li1±y(Mg1-xFex)P 的能带结构
图2为Li1±y(Mg1-xFex)P体系的能带结构图。由图2(a)(b)可知,自旋向上和自旋向下的子能带完全对称,且导带底和价带顶都在布里渊区的G点,故纯LiMgP为直接带隙半导体,带隙值为1.533eV,明显低于实验值Eg=2.43eV[18],这是因为计算方法中的DFT为基态理论,且GGA做的是近似处理,而能隙属于激发态,但这并不影响对Li1±y(Mg1-xFex)P电子结构及相关性质的分析[18-19]。图2(c)(d)为Li(Mg0.875Fe0.125)P能带结构图。Fe掺入后最显著的变化是在费米能级附近出现了均匀分布在自旋向上和自旋向下中的与Fe相关的10条自旋极化杂质带,杂质带宽度为0.726 eV,位于价带顶上方Ev+0.403 eV处,为受主能级,即在费米能级(EF)下方引入了空穴载流子,表明Fe掺杂LiMgP为p型掺杂。自旋向上的子能带表现出半导体性质,导带底由布里渊区的G点转变为Z点,表明体系由直接带隙半导体转变为间接带隙半导体,且带隙值增大为2.428 eV;自旋向下的子能带中杂质带跨过费米能级,故表现为金属性质,从而使整个体系表现出了半金属性质[26]。这种两个自旋子能带(自旋向上和自旋向下)分别呈现出半导体和金属特性的特殊能带结构可以产生自旋完全极化的传导电子,导致其在费米能级附近的自旋极化率(P)可以达到100%,明显表现为自旋注入,体系表现出半金属铁磁性.而半金属铁磁体的优劣程度可以用半金属能隙的大小来进行定量的分析[27],Fe掺杂LiMgP的半金属能隙为0.500eV(表2),明显大于Heusler合金中CrLiBi[28]的0.46eV和ZrFeVGa[29]的0.43eV,表明Fe的掺入使体系具有稳定的半金属性,在自旋电子器件领域具有较大的应用前景[30-32]。

图2 Li1±y(Mg1-xFex)P自旋极化能带图
图2(e、f)为Li1.125(Mg0.875Fe0.125)P的能带结构图。可以看出,由于此时体系中Li含量的增加,导致自旋向上的EF未贯穿在杂质带中,导带底由G点处的1.698eV转变到了Z点处的1.154 eV,价带顶由G点处的-0.739 eV降低到了-0.968 eV,故带隙值减小为为2.122 eV;自旋向下的EF贯穿在杂质带中,体系整体仍为半金属性,半金属能隙为0.095 eV,较单掺时明显减小,半金属性明显减弱。杂质带宽度较单掺时减小为0.348 eV,位于价带顶上方Ev+0.513eV处,EF向导带移动。图2(g、h)为Li0.875(Mg0.875Fe0.125)P能带结构,与Fe单掺和Li过量明显不同,Li不足时自旋向上与自旋向下的EF都贯穿于杂质带中,体系表现为金属性,导电性增强,杂质带宽度明显增大为1.268 eV。
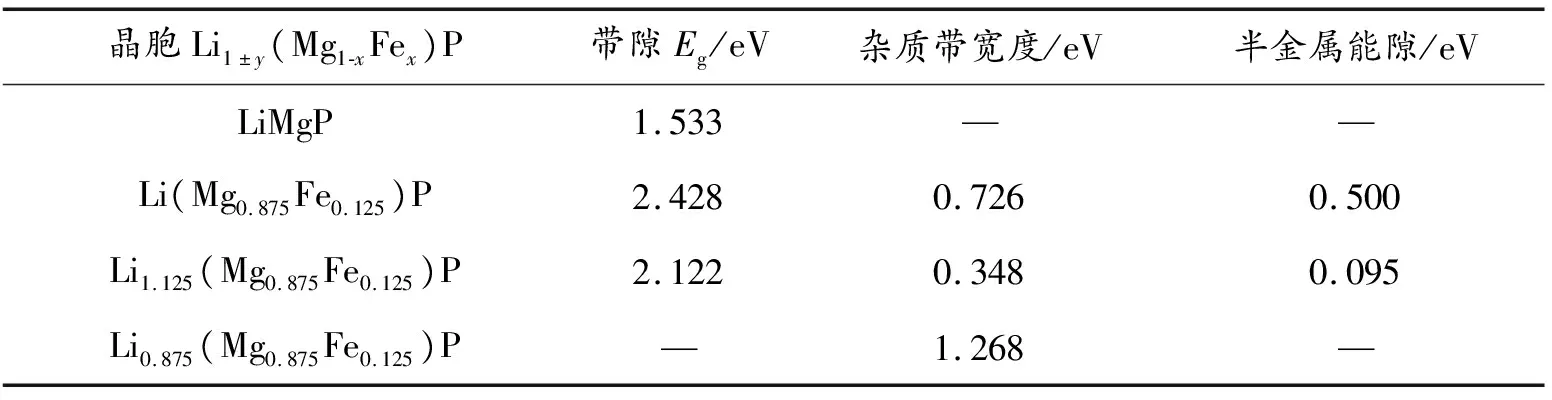
表2 Li1±y(Mg1-xFex)P带隙、杂质带宽度和半金属能隙
3 Li1±y(Mg1-xFex)P的态密度
图3为Li1±y(Mg1-xFex)P体系的态密度图。由图3(a)知纯LiMgP体系在费米能级附近的-2.6~0 eV上价带主要由Li2s、Mg2p和P3p电子构成;-4.3 eV~-2.6 eV之间的下价带主要由Li2s、Mg3s和P3p电子构成;导带底主要由Li2s和Mg2p电子构成,其中Li的2s态贡献最大。由图3(b)可知Fe掺杂LiMgP后,Li2s在价带和导带的态密度峰的峰值都明显减小,且在费米能级附近新出现的Fe3d态电子与Mg2p态电子的态密度的峰值出现交叠,表明Mg2p和Fe3d发生了p-d杂化作用,这种作用使得Fe3d态电子的t2g能级和eg能级处于劈裂状态,且部分t2g能级被推向费米能级之上,使其成为半填满状态,对费米能级以下的占据态进行积分计算,结果如表3所示,体系的净磁矩为4.02μB,其中Fe原子贡献最大,为3.48μB,Li、Mg 和P原子的贡献分别为-0.04μB,0.12μB,0.46μB。

表3 Li1±y(Mg1-xFex)P体系各原子磁矩和总磁矩.
从图3(c)可以看出,当Li过量时跨过费米能级的子带在Mg2p和Fe3d之外新增了Li2s,且Mg2p在费米能级附近的态密度峰值增大,而Fe3d在费米能级附近的态密度峰值减小,表明体系发生了sp-d轨道杂化作用,且轨道杂化的程度比单掺Fe的小,轨道劈裂程度减小,t2g能级向费米能级下方移动,体系净磁矩减小为3.06μB,主要来自于Fe原子贡献的2.94μB,Li、Mg和P原子的贡献分别为0μB,-0.10μB,0.22μB。由图3(d)可知,Li不足时跨过费米能级的自旋向上的子带主要为p3p和Fe3d,而自旋向下的子带主要为Fe3d和少量的Mg2p,表明体系自旋向上和自旋向下都发生了p-d杂化作用,体系整体上的轨道杂化作用增强,轨道劈裂程度增大,净磁矩增大为4.64μB, Li,Mg,P和Fe原子的贡献分别为-0.06μB,0.48μB,0.92μB,3.26μB。图3(e)(f)为Li1±y(Mg1-xFex)P体系的总态密度图,从图可得,Fe掺杂后体系态密度自旋向上和自旋向下不对称,体系具有净磁矩,这与表3计算结果相符合.
表4为Li1±y(Mg1-xFex)P体系的电子自旋净磁矩和电子自旋密度的模。2*integrated spin density代表电子自旋密度,即自旋向上电子数与自旋向下电子数的差值,它是以波尔磁子为单位的体系的自旋磁矩,2*integrated |spindensity|代表电子自旋密度的模。当二者不为0且相等时,表示电子自旋磁矩方向一致,体系具有铁磁性;当前者为0后者不为0时,表明相邻的电子自旋磁矩方向相反,体系具有反铁磁性;二者都为0时,体系为顺磁性[33]。为了确定掺杂模型的磁性,分别对Li1±y(Mg1-xFex)P体系自旋向上与自旋向下的电子密度进行定值计算。由表4可知,掺杂体系中2*integrated spin density和2*integrated |spindensity|值都不为0,且两者之间的差值很小,所以在误差允许范围内可视为近似相等,因此Li1±y(Mg1-xFex)P(x=0.125;y=0, 0.125)均为铁磁体,表明Fe的掺入能使LiMgP具有良好磁性。

图3 Li1±y(Mg1-xFex)P的分波态密度图。
表4Li1±y(Mg1-xFex)P体系的电子自旋净磁矩、电子自旋密度的模
Table4The2*integratedspindensity,2*integrated|spindensity|ofLi1±y(Mg1-xFex)P
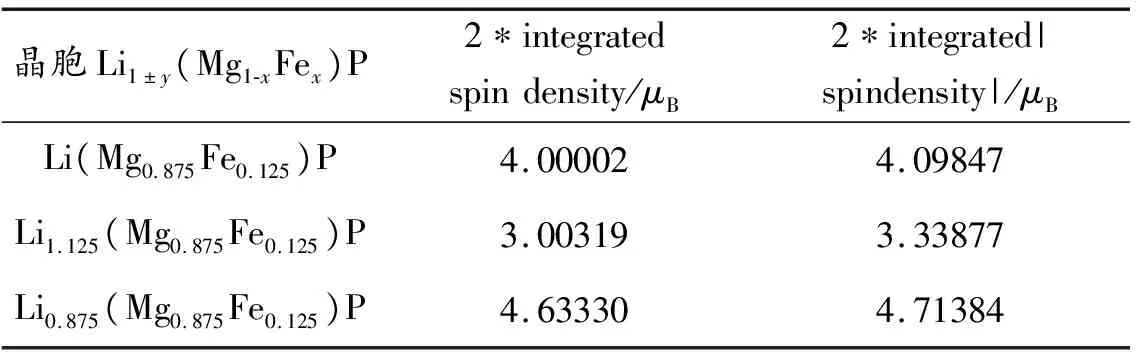
晶胞Li1±y(Mg1-xFex)P2∗integratedspindensity/μB2∗integrated|spindensity|/μBLi(Mg0.875Fe0.125)P4.000024.09847Li1.125(Mg0.875Fe0.125)P3.003193.33877Li0.875(Mg0.875Fe0.125)P4.633304.71384
4 Li1±y(Mg1-xFex)P的电荷布局、差分电荷密度和离子电子数
表5为Li1±y(Mg1-xFex)P体心原子与近邻原子之间的重叠电荷布局和键长的大小。LiMgP中的Li-P键和Mg-P键的重叠电荷布局全为正值,化学键表现为极化的共价键[34]。当Fe掺入后,Li-P键的重叠电荷布局变为-0.05,由共价键变为离子键。Mg-P键的重叠电荷布局增大为0.18,Fe与P原子间的电子发生了强烈的相互作用,使得Fe-P的重叠电荷布局为0.60,表明形成了强于Mg-P键的Fe-P共价键。在Li过量情况下,Li-P键电荷布局为0,表明Li、P之间不成键。Fe-P键的重叠电荷布局有所减小,由图3(c)可知,这是因为在Li过量体系中Fe3d在费米能级处的态密度值较单掺Fe时有所减小,且Fe3d轨道和其他原子轨道之间发生了相互作用,使得Fe-P键的相互作用有所减弱。此时Li-P键,和Fe-P键的键长都有所减小,而Mg-P键的键长有所增大,但整体上键长减小,与体系晶格常数减小相对应。Li不足时,Fe-P键的重叠电荷布局最大,从图3(d)可知Fe3d态自旋向下电子在费米能级处的态密度值较单掺Fe时减小,但是此时在费米能级处新增了自旋向上的Fe3d态电子,且P3p在费米能级处的态密度值明显增大,所以此时Fe3d轨道和P3p轨道之间的相互作用增强,使得Fe-P键的重叠电荷布局反而增大,增大为0.78,且键长最小,相互作用最强,使Li空位呈现金属性。
表5Li1±y(Mg1-xFex)P的键长、重叠电荷布局
Table5ThebondlengthsandoverlappingchargedistributionofLi1±y(Mg1-xFex)P

晶胞Li1±y(Mg1-xFex)P化学键重叠电荷布局键长/nmLiMgPLi-P0.030.26176Mg-P0.140.26093Li(Mg0.875Fe0.125)PLi-P-0.050.25315Mg-P0.180.26358Fe-P0.600.25993Li1.125(Mg0.875Fe0.125)PLi-P00.25310Mg-P0.210.26426Fe-P0.560.25153Li0.875(Mg0.875Fe0.125)PLi-P-0.020.24852Mg-P0.130.26958Fe-P0.780.22430
图4为4个体系晶面为(1, 1, 2)的差分电荷密度图,白色区域表示得电子,黑色色区域表示失电子。由图4(a)可知,P原子的第二层轨道被极化,电子发生内向移动,在图中表现为P原子中间存在一部分较小的白色区域[35]。P原子表现为得电子,虽然Mg的电负性强于Li,但由于Mg的电子多于Li的电子,因此体系靠近Mg位置的P原子的电子云却更密集,主要是Mg与P之间形成极化共价键且共用电子向P偏移所致。由图3(a)分波态密度图可知,共价键主要由Mg的2p态与P的3p态构成。从图4(b)可以看出,Fe与P之间为共价键,由于Fe的电负性强于Mg的电负性,故 Fe的掺入使共用电子偏移P原子的程度有所减弱,杂化作用增强,P原子周围的电荷分布仍存在方向上的极化分布,结合表5与图3(b)可知,主要是Mg的2p态电子、P的3p态电子、Li的2s态与Fe的3d态之间形成了极化共价键。图4(c)表明在Li过量情况下,较单掺Fe时,Fe原子和靠近它最近的P原子之间的电子云密集程度有所增强,且共用电子偏移程度有所减小,结合态密度图3(c)可知,因为此时Mg的2p态和Li的2s态电子在费米能级附近的值增大,Fe的3d轨道的部分电子在和P原子轨道发生相互作用的同时也要和Mg的2p轨道和Li的2s轨道发生相互作用,这使得Fe原子和P原子之间的相互作用最弱,重叠电荷布局的值最小。图4(d)表明Li不足时,Fe原子和靠近它最近的P原子之间的电子云最密集,且共用电子偏移程度最小,结合态密度图3(c)可知,Fe的3d态电子虽然在费米能级处的值有所减小,但是新增了自旋向上的一部分P的3p态电子在费米能级处的值明显增大,此时Fe的3d轨道和P的3p轨道之间的相互作用最强,重叠电荷布局的值达到最大,体系的磁性也最强,与单掺Fe相比,P原子的电子云存在部分缺失,这是由于Li原子的缺失使得P原子没有得到Li原子提供的电子所引起的,故在一定程度上解释了Fe与P电子云最密集的原因。

表6 Li1±y(Mg1-xFex)P体系中体心离子附近各离子的参数

图4 Li1±y(Mg1-xFex)P差分电荷密度图
表6为Li1±y(Mg1-xFex)P体心离子附近各离子的电参数。Li*表示的是体系中填隙处的Li离子,ns、np、nd和nt.分别表示的是s、p、d轨道上的电子数和总电子数,总电子数等于各个轨道上的电子数之和,charge表示的是离子的电荷数。由表6可知,本征LiMgP中Li离子在s轨道上的电子数和总电子数皆为2.08,电荷值为0.92,此时Mg离子nt的值为7.55,电荷数为0.45,P离子nt的值为6.37,电荷数为-1.37。Fe掺入后,Li离子的ns即nt减少为2.01,也就是Li离子轨道电子数又失去了0.07,所以Li离子电荷数增大为0.99。Mg离子的ns减小0.03,np减小0.02,故nt减小为0.05,Mg离子的电荷数增大为0.5。P离子的nt减少为6.24,主要是源于np减小了0.12,因此P离子也失去更多电子使得电荷数增大为-1.24,这与差分电荷密度图4(b)中P原子失去了更多电子的结果相符合。Li、Mg和P离子电子数减少是因为Fe掺杂之后,3种离子在原有相互作用的基础上,会有一部分电子和新增加的Fe离子之间发生轨道杂化作用,这种相互作用使得3种离子轨道上失去更多的电子,所带的电荷数增大,轨道电子数和总电子数减少可体现在态密度图3(b)中Li2态、Mg2s态、Mg2p态、P3s态和P3p态的态密度值都减小。Fe离子的nt为8.33,比其他离子nt都大的原因是因为Fe元素是过渡元素,所以具有的电子数最多,其中s、p、d轨道贡献的电子数分别为0.62、1.11、6.60。
Li过量时,Li离子的ns即nt减少为2.07,所以Li离子电荷数增大为0.93。Li*离子的ns为2.39,所带的电荷数为0.61,其轨道电子数大于Li离子的电子数是因为Li*离子是在原有体系的基础上填隙进去的,所以它和其他离子之间的相互作用相比其他位置的Li离子相对较弱。Mg离子和P离子的轨道电子数都有所减小,所以Mg和P离子之间的轨道杂化作用增强,重叠电荷布局增大,在费米能级处态密度值增大;Fe离子的轨道电子数增大,故Fe离子和其他离子之间的轨道相互作用减弱,重叠电荷布局的值减小。Li不足与单掺Fe相比,Li、Mg和P离子的s和p的电子数和总电子数都明显减少,离子所带的电荷数明显增大,Fe离子的电子数仅增大0.03,这表明在Li不足时参与轨道杂化作用的电子数最多,轨道之间的相互作用最强,导致Fe-P重叠电荷布局最大,体系中轨道电子数减少体现在态密度图3(d)中Li原子、Mg原子和P原子的Li2态、Mg2s态、Mg2p态、P3s态和P3p态的态密度减为最小.
5 结论
采用平面波超软贋势和广义梯度近似的第一性原理计算方法,对Fe掺杂LiMgP,Li过量和Li不足的超晶胞结构进行几何结构优化,计算并分析了系的电子结构、半金属性、态密度、电荷重叠布局及体系中心离子附近的电参数。结果表明,Fe的掺入能使Li1±y(Mg1-xFex)P具有较好的磁性,调节掺杂体系中Li的化学计量数可以实现体系电性和磁性的分离调控,且Fe单掺时具有大的半金属能隙0.500 eV,半金属铁磁性稳定,在自旋电子学器件领域中有较大的应用前景。Fe的掺入使得Fe3d态与Mg2p态发生p-d轨道杂化,产生4.02μB的净磁矩,主要源于Fe元素贡献的3.48μB,体系中轨道电子数减小,离子所带电荷数增大。Li过量时,体系中填隙位Li原子的引入使得轨道杂化作用减弱,净磁矩减小为2.72μB,仅仅表现为微弱的半金属性,带隙值减小为2.122 eV,此时体系中化学键键长减小,晶格常数减小,形成能最低,结构最稳定。Li不足时,杂质带宽度增大为1.268 eV,体系中轨道上的电子数明显减小,Fe3d轨道与P3p轨道杂化作用增强,净磁矩达到最大值4.64μB,表现为金属铁磁性,Fe-P键重叠电荷布局达到最大值0.78,键长达到最小值,Fe和P原子之间的电子云分布最密集且共用电子对偏移程度最小。
猜你喜欢
无机化学学报(2022年9期)2022-09-16
中外文摘(2021年7期)2021-04-23
发明与创新(2020年39期)2020-10-15
复旦学报(医学版)(2020年3期)2020-06-18
原子与分子物理学报(2020年5期)2020-03-17
数学物理学报(2019年6期)2020-01-13
发明与创新·小学生(2019年12期)2019-12-05
中学生数理化·高三版(2017年1期)2017-04-20
中国塑料(2016年1期)2016-05-17
读写算·教研版(2016年8期)2016-05-07
