
利用CRISPR/Cpf1技术构建HEK293细胞DDX21基因稳定敲除株及功能鉴定
2020-03-12 00:40鲁国涛曾为俊邵玉乐陈洪岩孟庆文中国农业科学院哈尔滨兽医研究所兽医生物技术国家重点实验室黑龙江省实验动物与比较医学重点实验室黑龙江哈尔滨150069
中国兽医学报 2020年2期
鲁国涛,王 辉,曾为俊,邵玉乐,许 曼,陈洪岩,孟庆文 (中国农业科学院 哈尔滨兽医研究所 兽医生物技术国家重点实验室/黑龙江省实验动物与比较医学重点实验室,黑龙江 哈尔滨150069)
DEx D/H-box家族蛋白是一类重要的RNA 解旋酶,存在于几乎所有的真核生物和多数原核生物中。DEx D-box 解旋酶21(DEx D-box helicase 21,DDX21)是由FLORES-ROZAS 等[1]首次从HeLa细胞的核提取物中鉴定的解旋酶,研究证实,其不仅参与m RNA 前体加工、RNA 的转运和降解、核糖体的生成等多种生物学过程[2-3],而且在肿瘤的发生发展、参与机体的天然免疫应答[4-5]、调控A 型流感病毒(influenza A virus,IAV)、人类免疫缺陷病毒(human immunodeficiency virus,HIV)和登革热病毒(dengue virus,DENV)等多种病毒的增殖及信号传导中也起到非常重要的作用[6]。
禽流感病毒(avian influenza virus,AIV)属于正黏病毒科(Orthomyxoviridae)中的A 型流感病毒属。流感病毒感染过程中,多种模式识别受体能够识别病毒的RNA,活化转录因子NF-KB、诱导肿瘤坏死因子-α(TNF-α)、白介素-8(IL-8)等促炎症因子和Ⅰ型干扰素的产生,参与机体抗病毒免疫反应[7]。而在甲型流感病毒感染过程,DDX21可以与TRIF-S100A9-TLR4-My D88形成复合物进行信号传导,调节病毒感染过程中的促炎反应过程[8]。但是,DDX21在流感病毒信号传导及病毒复制过程中发挥的具体作用并不十分清楚。
本研究利用CRISPR/Cpf1基因编辑技术构建DDX21基因编辑细胞,通过基因测序、Western blot和H9N2亚型AIV 感染试验验证敲除DDX21基因的HEK293 细胞株功能变化,为深入研究DDX21基因功能、病毒感染的天然免疫应答和调控研究奠定基础。
1 材料与方法
1.1 材料HEK293 细胞、pcDNA3.1-h LbCpf1-eRFP质粒由本实验室保存;p U6-Lb-cr RNA 载体由哈尔滨工业大学黄志伟教授惠赠;H9N2 亚型AIV 来自哈尔滨兽医研究所保藏中心。
1.2 试剂Premix Ex Taq酶、DNA Marker、反转录试剂盒购自TaKaRa公司;PCR purification Kit、胶回收试剂盒购自Axygen 公司;Lipofectamine 2000购自Invitrogen 公司;DMEM 培养基和胎牛血清均购自Gibco 公司;各种限制性核酸酶购自NEB公司;细胞基因组DNA 提取试剂盒、质粒提取试剂盒购自TLANGEN 公司;β-actin抗体、DDX21抗体购自Omnim Abs公司;引物由哈尔滨博仕生物技术有限公司合成。
1.3 靶点选择及载体构建在NCBI 中检索DDX21基因(Gene ID:9188),利用网络工具chopchop(http://chop chop.cbu.uib.no/)设计g RNA序列;选择针对DDX21 基因特异性高的靶点(表1),分别在编码链的5′端添加AGAT,非编码链的5′端添加AAAA,与BsmBⅠ酶切p U6-Lb-cr RNA后形成的黏性末端互补。根据靶点位置设计相应的测序引物及鉴定引物,产物大小分别为245,612 bp(表1)。
gRNA 寡核苷酸单链程序性退火形成双链:取等量的上游链和下游链混合(终浓度为100μmol/L)进行退火,退火程序:95℃10 min,-1℃/s降至25℃。T4连接酶连接线性PU6-Lb-cr RNA 和退火产物,连接产物转化DH5α感受态细胞,挑取单菌落,提取质粒测序验证。
1.4 细胞的培养及转染用含10%胎牛血清的DMEM 培养基传代HEK293 细胞,将细胞浓度为5×105个HEK293细胞接种至6孔板中培养,待细胞生长达到50%~60%单层时,用转染试剂3μL及gRNA 表达载体质粒0.5μg、pcDNA3.1-h Lb-Cpf1-eRFP质粒1μg共同转染HEK293细胞,恒温培养箱中培养。

表1 DDX21-gRNA 及检测引物寡核苷酸序列
1.5 细胞基因组DNA的提取细胞转染12 h后,
加入1 mg/L G-418(geneticin,遗传霉素)筛选。在药物筛选48 h 后,取部分细胞利用细胞基因组DNA 提取试剂盒提取细胞DNA;取另一部分细胞用200目滤网过滤,流式细胞分选仪(BD 公司)分选表达红色荧光蛋白的HEK293细胞,96孔细胞培养板中进行细胞克隆化。
1.6 T7E1 分析检测T7E1 试验分析步骤:用PCR purification Kit试剂盒纯化PCR 产物,纯化后的产物退火并测定浓度,用T7E1酶切鉴定gRNA的打靶效率。2%琼脂糖凝胶电泳后,利用Image J计算条带灰度值,并通过公式计算切割效率:Indel(%)=100×[1- (1-fcut)1/2],fcut=(b+c)/(a+b+c),其中Indel为插入缺失比率,fcut为切割比率,a为未被切割条带的灰度值,b和c表示切割产生的新条带的灰度值[10]。
1.7 筛选稳定敲除DDX21基因的HEK293细胞株
分选的单克隆细胞传至12孔板,选取6株细胞提取基因组DNA,经PCR 扩增后测序与野生型(WT)基因组对比。选取成功造成DDX21基因移码突变的细胞,部分细胞用于传代,另一部分与正常细胞培养同时提取蛋白,通过Western blot检测蛋白表达。
1.8 遗传稳定性与细胞增殖速度分析将敲除组及WT 细胞以2×105个细胞接种于6孔板中,细胞连续传代后,提取基因组DNA,使用测序引物进行PCR 扩增;连接转化后,挑取单菌落送公司测序以鉴定遗传稳定性。显微镜下观察每5代敲除细胞株与WT 细胞株形成细胞单层的时间,每个细胞株做3次平行试验并进行统计分析细胞增殖速度。
1.9 细胞接毒H9N2 亚型AIV(HA 为1∶128,TCID50=10-5.5),以MOI=1 的量接种细胞,37℃培养箱中培养,6,12,18,24,36 h 不同时间点收取上清,检测TCID50,绘制病毒在HEK293细胞上的生长曲线。
1.10 模式识别受体、细胞因子及抗病毒蛋白mR-NA表达水平检测在不同时间点收取WT 对照组及敲除组接毒细胞上清,每个样品设置3个平行样本,提取RNA,反转录成cDNA;荧光定量PCR 检测TLR-3、TLR-7、MDA-5、IFN-α、IFN-β、IL-6 和OAS基因表达量。以GAPDH 为内参基因,采用2-ΔΔCt法进行计算,荧光定量PCR 引物参考文献[9](表2)。

表2 HEK293细胞荧光定量PCR 引物
1.11 统计分析利用Graphpad prism 6.0软件进行试验数据统计分析,Tukey’s test用于各组数据差异分析,P<0.05(∗)表示差异显著;P<0.01(∗∗)表示差异极显著。
2 结果
2.1 DDX21-gRNA 重组载体构建通过退火杂交形成的oligo二聚体与p U6-Lb-crRNA质粒线性载体连接,并将构建完成的质粒送吉林库美生物公司测序,命名序列正确的质粒为DDX21-gRNA(图1)。
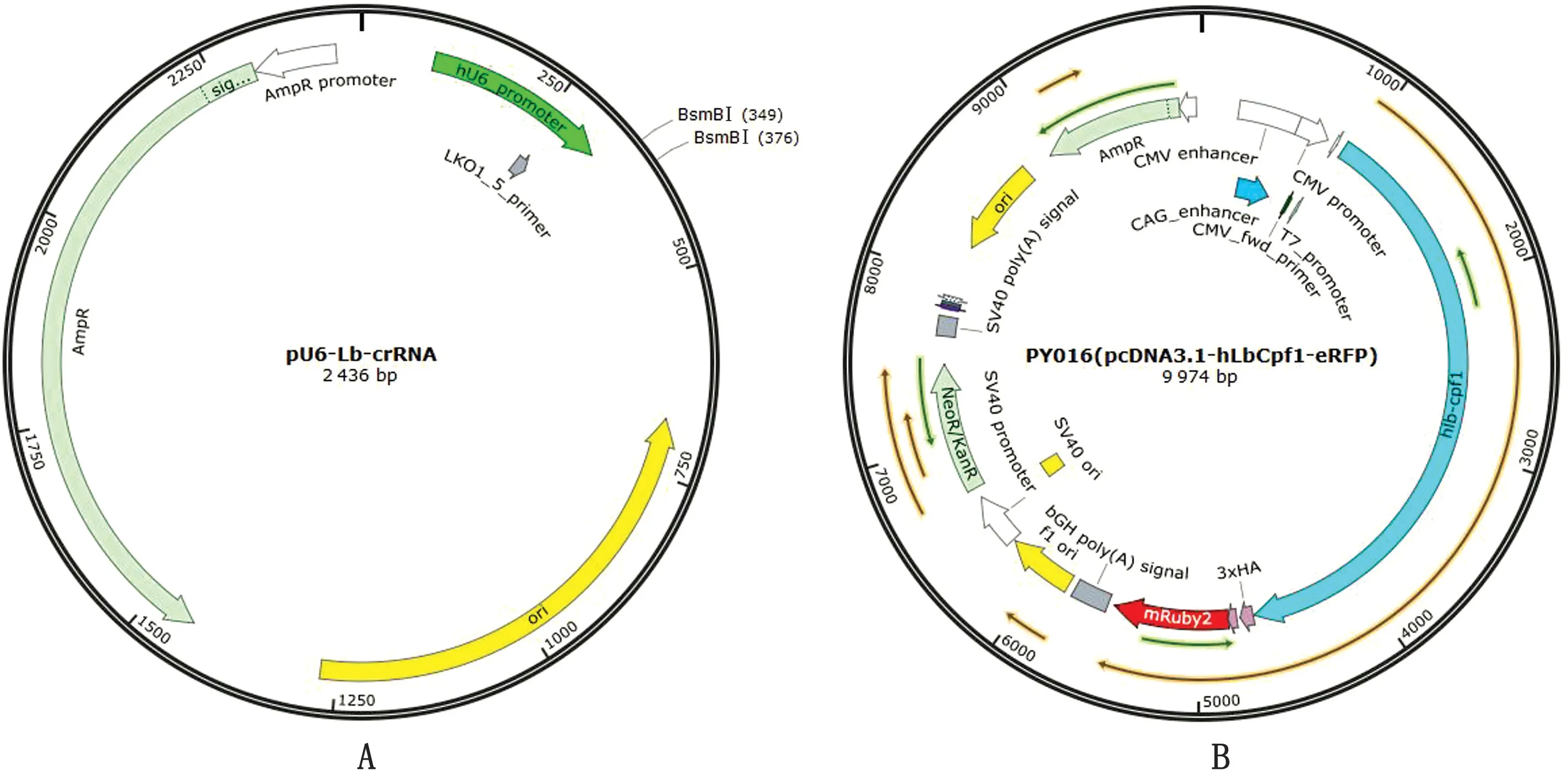
图1 CRISPR/Cpf1系统载体图谱 A.DDX21-gRNA;B.pcDNA3.1-h LbCpf1-eRFP
2.2 双质粒转染细胞共转染HEK293细胞,在倒置荧光显微镜下观察,表达红色荧光蛋白,表明质粒转染HEK293细胞成功(图2)。
2.2 T7E1检测gRNA活性用T7E1酶切,经2%琼脂糖凝胶电泳分析g RNA 有较高的打靶效率;Image J软件计算灰度值,通过公式计算gRNA 活性约为56.8% (图3)。
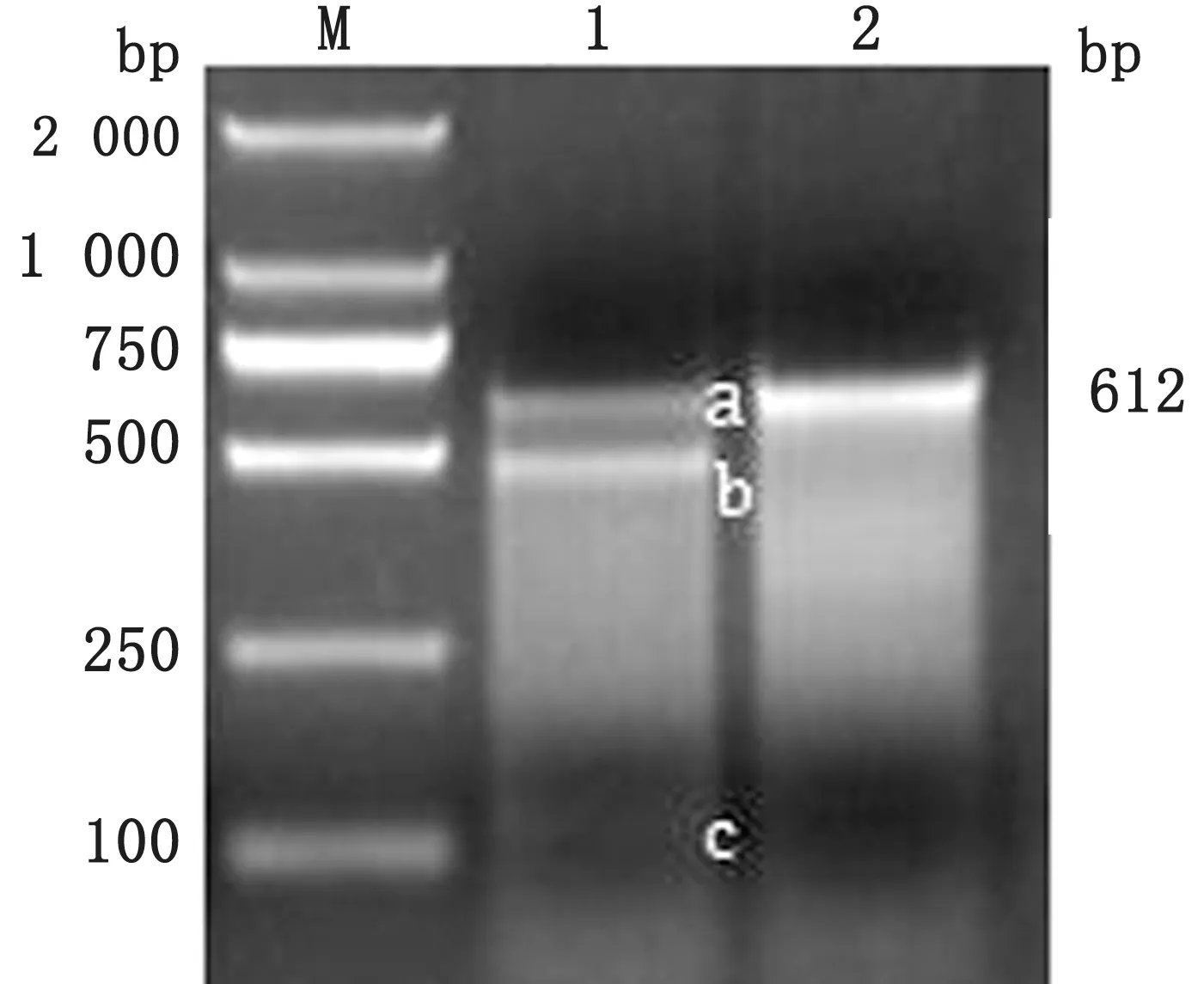
图3 T7E1检测gRNA 切割效率 M.DL2000 DNA Marker;1.T7E1分 析DDX21-g RNA 有 效 性;2.阴 性 对 照。a.未被切割条带;b,c.分别表示切割产生的新条带
2.4 DDX21基因敲除细胞株的建立测序结果:6株细胞中有2个单克隆细胞株的2条等位基因在靶点附近分别缺失16,26 bp,下划线序列为所设计g RNA,加 粗 字 体 为PAM 序 列。WT:5′-AGGCAGTAGAGGCCCGAGAGGACAGCGATCAGGAGGTGGCAACAAA-3′;-16 bp 5′-AGGCAGTAGAGGCCCGAGAG…… …… ……TGGCAACAAA-3′;-26 bp 5′-AGGCAG………………GAGGTGGCAACAAA-3′。
2.5 Western blot检测敲除细胞中DDX21蛋白表达对敲除细胞及WT 细胞提取总蛋白,Western blot检测结果显示DDX21蛋白不表达,证明成功获得2株敲除DDX21基因的HEK293细胞株(图4)。
2.6 DDX21基因敲除细胞系遗传稳定性及增殖速度分析连续传代后测序鉴定2株敲除细胞株基因仍缺失,证明2株敲除DDX21基因的细胞株能够稳定遗传。对不同代次敲除与WT 细胞形成细胞单层时间进行统计分析,结果表明,敲除组与WT 组相比形成细胞单层的时间明显延长,差异具有统计学意义(P<0.01),提示DDX21可能参与细胞的增殖(图5)。
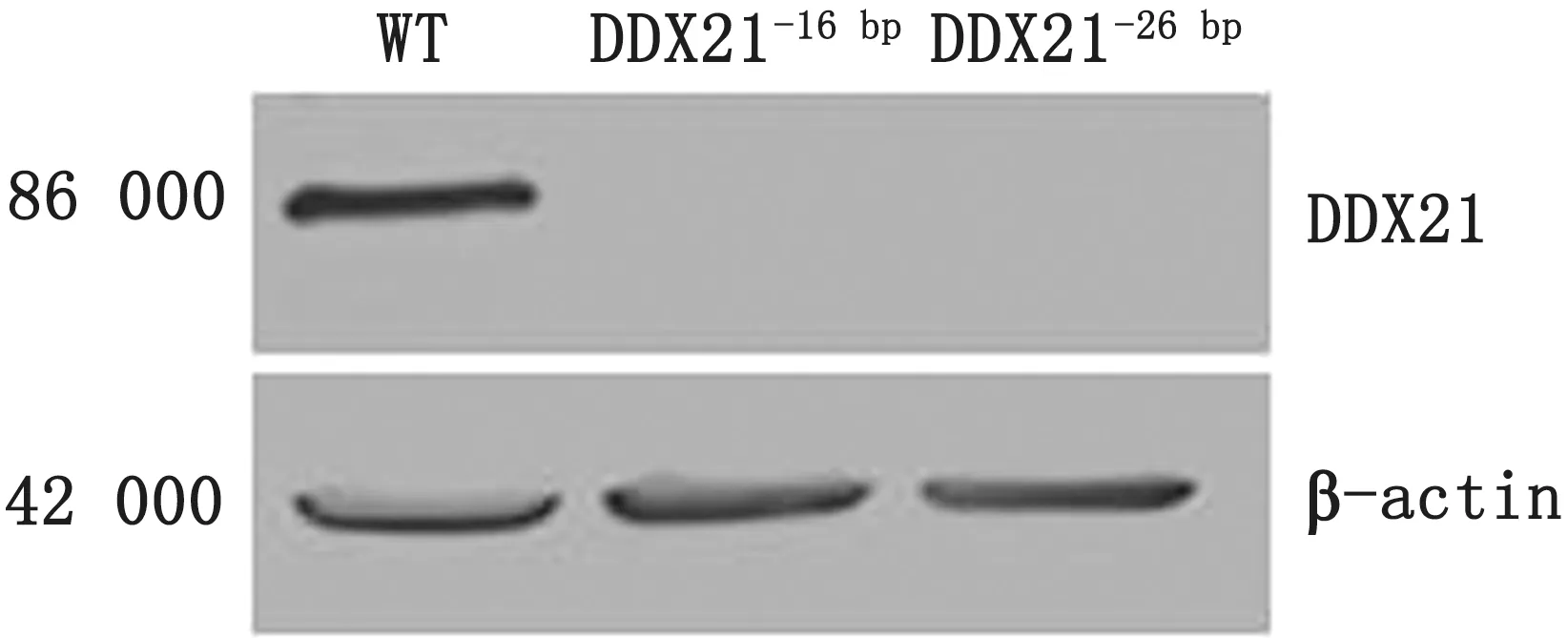
图4 Western blot检测DDX21-gRNA 的靶向敲除效果
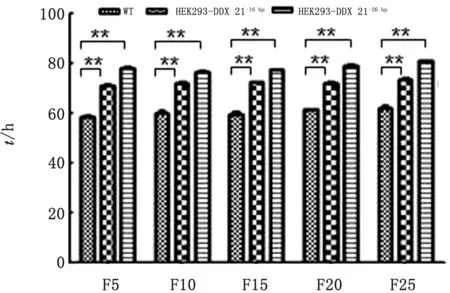
图5 野生型与基因敲除细胞形成细胞单层时间
2.7 病毒生长曲线由图6可知:病毒感染早期复制较快,6~36 h变化范围为1.17~3.01倍,其中在24 h达到峰值(P<0.01),36 h时敲除组与对照组差异显著(P<0.05)。结果表明:HEK293-DDX21-26bp细胞显著提高了流感病毒产量。
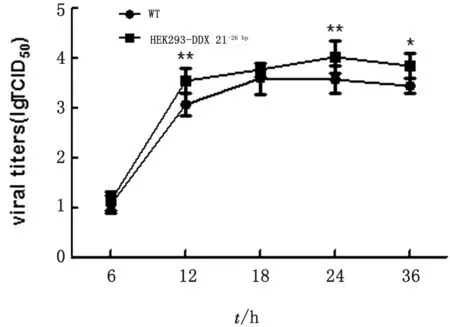
图6 病毒在HEK293细胞上的生长曲线
2.8 模式识别受体、细胞因子及抗病毒蛋白mRNA
表达水平检测荧光定量PCR 检测模式识别受体、细胞因子及抗病毒蛋白m RNA 表达水平,结果显示:敲除组TLR3 m RNA 表达水平在6~12 h低于对照组,18 h时达到峰值(P<0.01)(图8A);TLR-7基因表达变化不明显(图8B);MDA-5表达水平为对照组的2.04倍(P<0.01)(图8C);而OAS m RNA 及细胞因子IFN-α、IFN-β表达水平明显低于对照组(P<0.01)(图8D~F);IL-6呈先升高后降低趋势,但表达水平低于对照组(图8G);表明DDX21基因敲除阻断DDX21-TRIF-MyD88信号通路,抑制Ⅰ型干扰素、炎症因子和抗病毒蛋白表达。
3 讨论
目前文献报道有5种基因编辑方法:同源重组技术、锌指核酸酶技术、转录激活因子样效应物核酸酶技术、CRISPR/Cas9 基因编辑技术和CRISPR/Cpf1基因编辑技术[10]。其中CRISPR/Cpf1 基因编辑技术是2015 年报道的一种新的基因编辑技术[11]。本研究选用CRISPR/Cpf1 进行基因编辑,因为该方法不仅有较好的切割活性,而且具有较高的特异性[12]。研究中利用的pcDNA3.1-h LbCpf1-eRFP载体同时带有红色荧光报告基因和G-418药物筛选基因,红色荧光蛋白的表达不仅能够指示质粒转染细胞的效率,同时也为G-418药物筛选提供了选择的时机。而通过药物筛选存活的细胞基本都为CRISPR/Cpf1靶向的阳性细胞,报告基因和药物筛选基因的同时选择,大大提高了靶向敲除DDX21基因的成功率,减少了后续阳性细胞的筛选工作。本研究利用CRISPR/Cpf1 系统成功获得了2 株DDX21基因稳定敲除的HEK293 细胞系,沉默了DDX21基因的表达,为研究DDX21的作用机制提供了理想的细胞模型,同时为进一步研究其他哺乳动物细胞DDX21基因敲除提供了成功的技术路线和可操作的试验方法。
HENNING 等[13-14]的研究发现沉默细胞中DDX21 基因表达,能够抑制细胞中18S 和28S RNA 产生并减缓细胞增殖,提示DDX21可能与细胞生长和增殖因子相互作用,参与细胞的增殖过程。本研究获得的敲除细胞株连续传代后,测序结果表明敲除细胞株具有良好的遗传稳定性。与对照组相比,敲除组形成细胞单层的时间明显延长(P<0.01),表明敲除DDX21基因可影响细胞的增殖速度。
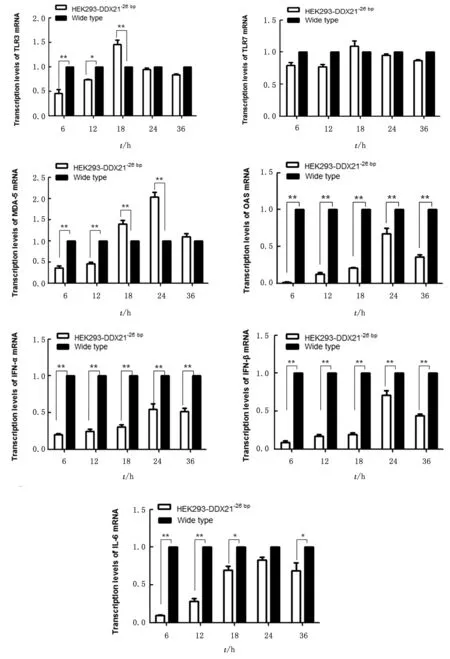
图8 H9N2亚型AIV 感染后HEK293-DDX21-26 bp细胞系中TLR-3、TLR7、MDA-5、OAS、IFN-α、IFN-β及IL-6 m RNA 变化
流感病毒感染后,DDX21 与下游接头蛋白TRIF相互作用,促进S100 蛋白家族基因S100A9表达,并激活TLR4-My D88途径[8]。通过荧光定量PCR 结果显示,敲除DDDX21基因后,模式识别受体TLR-3、MDA-5 的m RNA 表达水平上调,IFNα、IFN-β、IL-6及抗病毒蛋白OAS的m RNA 表达水平下调;表明敲除DDX21基因能够阻断DDX21-TRIF-MyD88信号通路,抑制下游IFN-α、IFN-β、OAS及IL-6 的表达,引起上游TLR-3 和MDA-5的累积。
本研究利用CRISPR/Cpf1 技术构建敲除DDX21基因的HEK293 细胞株,通过TA 克隆测序、Western blot和H9N2亚型AIV 感染后的免疫反应对其进行验证,获得DDX21 基因稳定敲除的HEK293细胞株,为揭示DDX21参与IAV 感染引发的天然免疫应答及作用机制研究提供理论依据。
猜你喜欢
传染病信息(2021年6期)2021-02-12
科海故事博览·下旬刊(2019年6期)2019-04-16
食品科学(2018年10期)2018-05-23
西南医科大学学报(2015年1期)2015-08-22
医学研究杂志(2015年11期)2015-06-10
中国当代医药(2015年16期)2015-03-01
中国当代医药(2015年9期)2015-03-01
中国医药导报(2015年27期)2015-02-28
生物医学工程学进展(2015年1期)2015-02-28
化学工业与工程(2015年1期)2015-02-10
