
Distribution of Protists in the Deep South China Sea Revealed by High-Throughput Sequencing
2020-03-09 04:52LIXinranWARRENAlanJIAONianzhiandXUDapeng
LI Xinran, WARREN Alan, JIAO Nianzhi, and XU Dapeng, *
Distribution of Protists in the Deep South China Sea Revealed by High-Throughput Sequencing
LI Xinran1), WARREN Alan2), JIAO Nianzhi1), and XU Dapeng1), *
1),,,,361102,2),,
Protists (microbial eukaryotes) are indispensable members of the marine microbial food web. In recent years, organisms living in the deep sea (>1000m water depth) have increasingly become the focus of research; however, studies on protistan assemblages are relatively scarce compared with their prokaryotic counterparts. In the present study, high-throughput sequencing of the hypervariable V9 region of the 18S rRNA gene was used to explore the community composition of protists in bathypelagic waters of the South China Sea. Based on the analysis of the alpha and beta diversities of 14 samples, we discovered: 1) members belonging to Rhizaria, Alveolata, and Excavata were the dominant groups in terms of both relative sequence abundance and operational taxonomic unit (OTU) richness in all samples, although their relative contributions differed among different samples; 2) cluster analysis showed that the distribution of protistan assemblages was related neither to the sampling location nor to the water depth, and other environmental factors might have caused the differences among the communities; 3) phototrophs, including members of the Bacillariophyta, Bolidophyceae, Dictyochophyceae, Prasinophyceae, and Prymnesiophyceae, were detected in all samples, which indicated their contributions to the downward transportationthe biological pump and the potential presence of phagotrophy of these phototrophic cells in the deep ocean.
bathypelagic water; diversity; microbial eukaryotes; SSU rRNA gene
1 Introduction
Marine protists (microbial eukaryotes) are highly diverse (both morphologically and phylogenetically) unicellular eukaryotes that occur in almost every branch of eukaryotic evolutionary tree (Bass., 2005; Guillou., 2008; Song, 2009; Caron., 2012; Sun, 2017; Adl, 2019; Hu, 2019). Their size range spans more than four orders of magnitude (Caron, 2009). The nutritional modes of protists include phototrophy, heterotrophy, and mixotrophy, and they serve as primary producers, consumers, decomposers and trophic links in aquatic food webs which are essential for the biogeochemical cycles of the ocean (Azam., 1983; Jiao., 2010; Caron., 2017).
The deep dark ocean is divided into three parts: the mesopelagic zone (200–1000m depth), the bathypelagic zone (1000–4000m depth), and the abyssal zone (>4000m depth). It is considered to be the largest habitat on Earth and is characterized by high inorganic nutrient con- centrations, low temperature, high pressure, and is the world’s largest reservoir of dissolved organic carbon (Arístegui, 2009; Herndl and Reinthaler, 2013). Due to the expense and technical difficulties in sampling and performingexperiments, both prokaryotic and eukaryotic microbes living in the deep waters are understudied compared with the microbes living in the sunlit ocean (Arístegui., 2009). Fortunately, the culture- independent techniques such as denaturing gradient gel electrophoresis (DGGE), clone library, and high-through- put sequencing using marker genes (., the small subunit ribosomal DNA) have provided powerful methods for studying microbial diversity in the deep ocean (Arís- tegui., 2009). Nevertheless, deep sea protists have been largely neglected compared with their prokaryotic counterparts (bacteria and archaea), leaving their diversity and community composition largely unresolved. Studies on deep sea protists both on local and global scales have increased significantly in recent years, uncovering their new clades, distribution patterns and potential ecological roles (Countway, 2007; Not, 2007; Pernice, 2015; Xu, 2017a, b). However, given the vast area of the deep sea environment, much work needs to be done in order to reveal the biodiversity of protists and the roles they play in biogeochemical cycles in the deep sea.
In the present study, high-throughput sequencing of the hyperviable V9 region of the SSU rRNA gene was performed to investigate the community composition of protists from the bathypelagic waters of the South China Sea, one of the largest marginal seas in the world.
2 Materials and Methods
2.1 Sample Collection
Six sampling stations were chosen to represent the central part of the deep basin in the South China Sea (SCS) (Fig.1). Seawater samples were collected using Niskin bottles attached to a CTD rosette system aboard thefrom 1000m depth at all stations, five samples were taken from 2000m depth except at station H7, and three samples were taken at 4000m depth at stations H7, O3 and O14, respectively. For each sample, two liters of sea water were prefiltered through 200µm Nitex (Sefar) mesh and then collected by polycarbonate filters with a pore- size of 0.22µm (Millipore). The filters were immediately frozen in liquid nitrogen and stored at −80℃ until further treatment.
Fig.1 Geographic locations of the sampling sites in the central South China Sea. The map was generated using Ocean Data View 4 software (Schlitzer, 2011).
2.2 DNA Extraction, PCR Amplification, and High- Throughput Sequencing of SSU rRNA Gene
Genomic DNA was extracted from each sample using the Power-Water DNA Isolation Kit (MoBio Laboratories, Inc., Carlsbad, CA) following the protocols from the manufacturer with minor modifications (Sun, 2017; Xu, 2017a). Extracted DNA was stored at −20℃ for later processing. Universal primers 1389F (5’-TTGTACA CACCGCCC-3’) and 1510R (5’-CCTTCYGCAGGTTC ACCTAC-3’) were used to amplify the hypervariable V9 region of the SSU rRNA gene (Amaral-Zettler, 2009). Six individual PCR reactions for each sample were run employing Ex Taq DNA polymerase (TaKaRa, Dalian, China) and the products were pooled to collect sufficient amplicons for sequencing. The pooled PCR products were purified using Wizard®SV Gel and PCR Clean-Up System (Promega, Beijing, China). Bridge amplification and paired end sequencing of the amplicons were performed with an Illumina MiSeq platform by a commercial sequencing company. Sequence data generated have been deposited in the NCBI Sequence Read Archive with accession number SRP104547.
2.3 Sequence Analysis
Quality filtering, demultiplexing and assembly of raw data were conducted with Trimmomatic (Bolger, 2014) and Flash software (Magoč and Salzberg, 2011) and criteria employed followed Li(2018). Potential chimeras were identified and removed using UCHIME (Edgar, 2011) applying bothand reference- based chimera searches against the Protist Ribosomal Database 2 (PR2) (de Vargas, 2015). Singletons (reads present as a single copy) were removed from downward analysis. Operational taxonomic units (OTUs) were clustered at a 95% similarity threshold using UPARSE (Edgar, 2013). Taxonomy assignment of each OTU were achieved using BLAST method against PR2 as implemented in QIIME (Caporaso, 2010). Non-protistan OTUs (., bacteria, archaea, metazoa, fungi, plastids, or OTUs classified as ‘Unassigned’) were removed.
All datasets were sub-sampled at a uniform depth of 13842 sequences (the lowest sequence count for all samples) before downstream analysis. An OTU table was generated and alpha diversity indices (Chao1, Shannon, ACE, Inverse of Simpson, and Phylogenetic Diversity) were calculated using QIIME (Caporaso, 2010). Non-metric multidimensional scaling (NMDS) was constructed using the Vegan package as implemented in R based on the Bray-Curtis coefficient. Weighted Unifrac metric, which compares samples based on the phylogenetic relatedness of OTUs in a community and considers relative OTU abundance, was used to infer the grouping of samples (Lozupone and Knight, 2005).
3 Results
To infer the diversity of protistan assemblages in the bathypelagic waters of the central South China Sea, a total of 14 samples from 6 stations were collected (Fig.1). After quality-screening, a total of 857544 sequences with an average length of 123bp (mean±std: 123.0545±16.4996) were generated. After removal of potential chimeras, singletons, and sequences that were not affiliated with protists, there were a total of 366951 sequences, ranging 13842 to 45271 reads per sample (Table 1). All datasets were rarefied at a uniform depth of 13842 sequences (the lowest number of sequences for all samples). The final OTU table containing 1911 OTUs and 193788 sequences was used to analyze alpha and beta diversities and taxonomic affiliations of bathypelagic protists.
3.1 Alpha and Beta Diversities of Bathypelagic Protists
Alpha diversity estimates are shown in Table 1. The richness observed varied among samples, ranging from 927 to 1577 OTUs per sample. The average Shannon index was 5.57±0.40,while the highest one (7.24) was found at 2000m deep waters of station O1 and the lowestone was at 2000m deep waters of station O14. Phylogenetic diversity (PD), which measures the total branch length connecting all OTUs in the SSU rRNA gene phylogeny, ranged from 213.5 (1000m deep waters at station O1) to 137.9 (2000m deep waters at station O7) (Table 1). Rarefaction curves showed that all samples were not fully sampled (Fig.2A). However, a rarefaction curve for the pooled dataset from all samples showed a symbol saturation that a maximal richness of bathypelagic protists was approximately 2000 OTUs (Fig.2B).
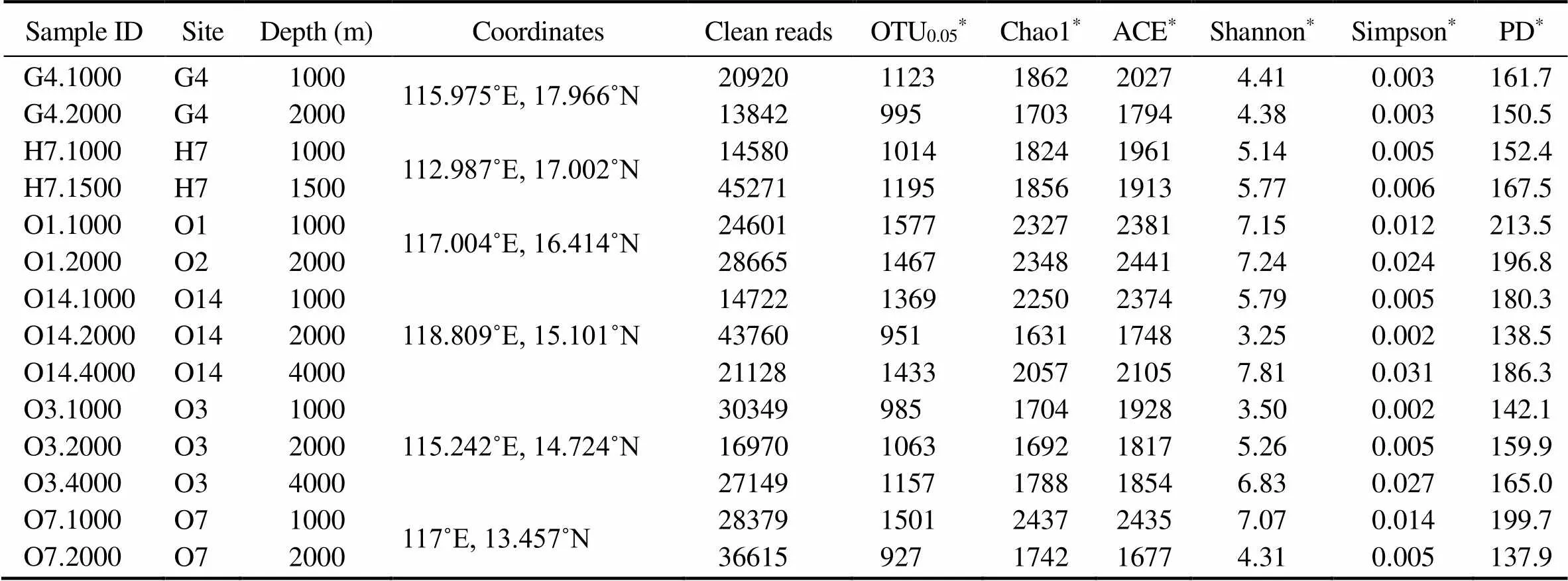
Table 1 Diversity estimates of South China Sea samples
Notes: OTU0.05, Operational taxonomic unit at 95% 18S rRNA V9 gene sequence identity.*Standardized numbers based on subsampling of 13842 sequences without replacement.
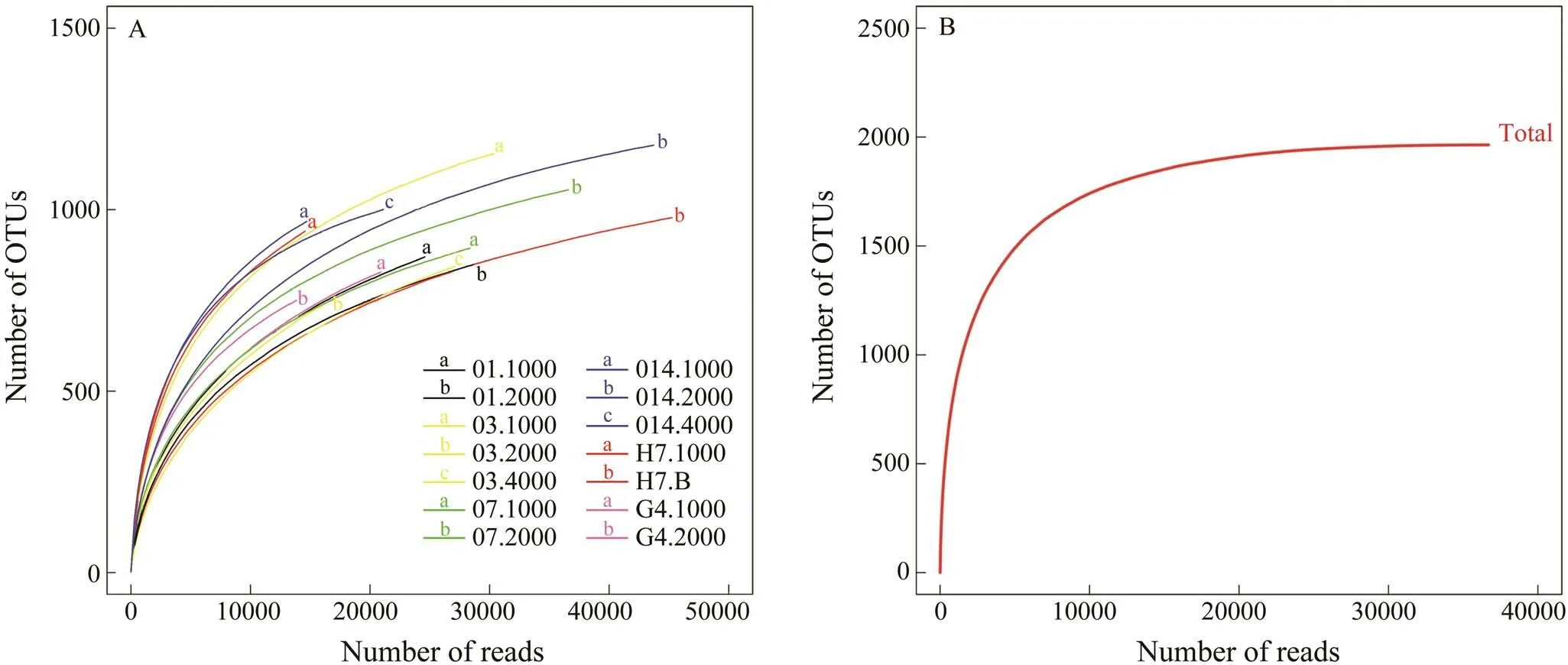
Fig.2 Alpha diversity of protists in bathypelagic waters of the South China Sea as inferred by clustering reads at 95% similarity. (A) Rarefaction curves for each sample. (B) Global rarefaction curve.
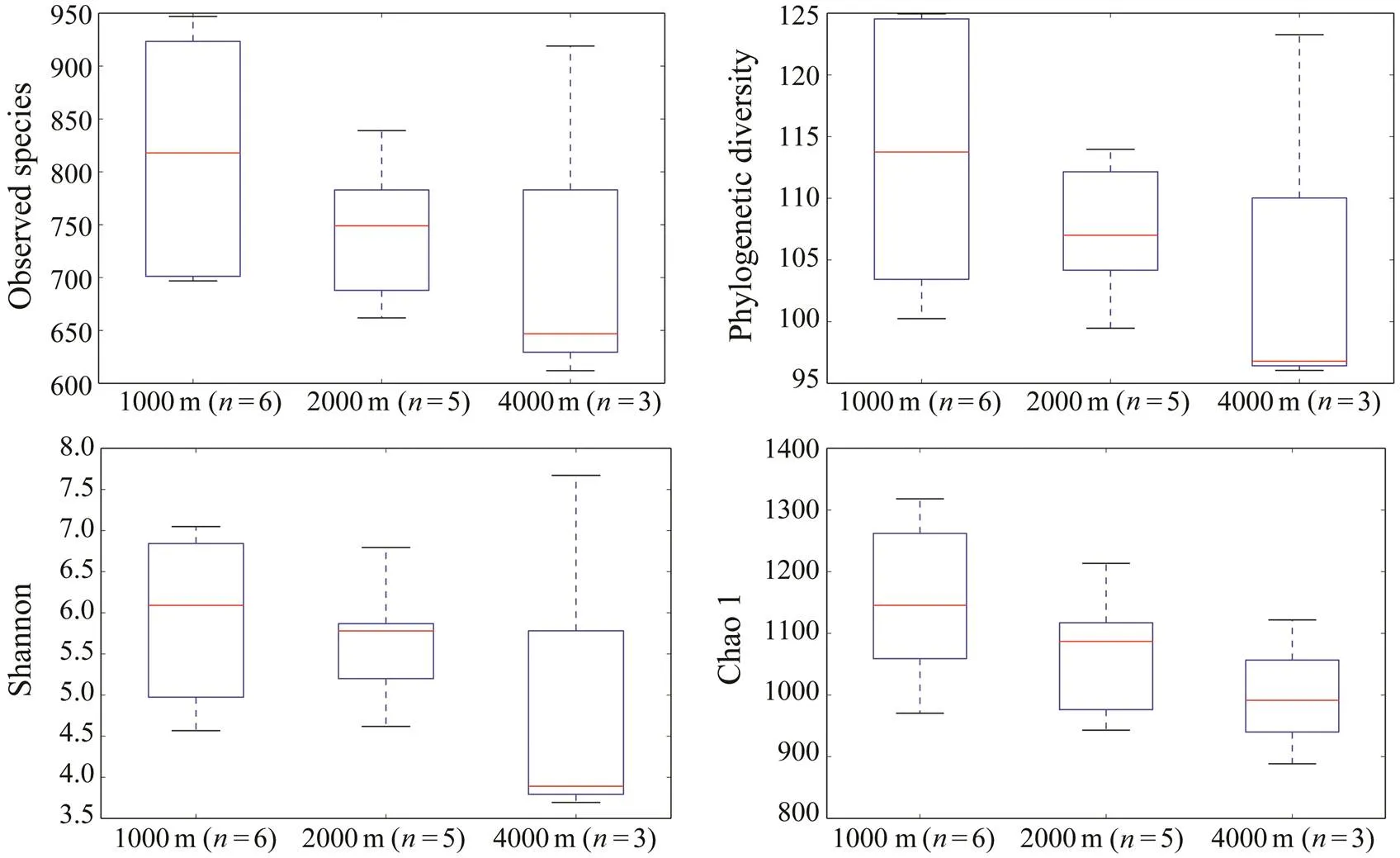
Fig.3 Alpha diversity measures (Observed species, Phylogenetic Diversity, Shannon and Chao1) for the pooled samples grouped by water depths. The box delimits the 25th and 75th percentiles.The whisker shows the range and the line in every box plot indicates the median.
To reveal the differences of alpha diversity at different depths, samples retrieved from the same depth were pooled and the nonparametric test (Monte Carlo permutations) was conducted based on the Observed species, Phylogenetic diversity, Shannon and Chao1. As a general trend, the average alpha diversity decreased as water depth increased although there was no significant difference among sample groups (Fig.3).
A non-metric multidimensional scaling ordination (NMDS) analysis, based on Bray-Curtis similarity, was implemented to explore the community composition of bathypelagic protists among all 14 samples. The samples were clustered into three groups. Group 1 contained samples O14.4000 and G4.2000; group 2 contained samples O14.1000, O3.1000, and H7.1000; and group 3 contained the rest of the samples (Fig.4A). This grouping pattern was also supported by the principle component analysis (PCoA) of community taxonomic relatedness quantified by the Weighted Unifrac metric (Fig.4B). Statistical ana- lyses showed the composition of the three groups was sig- nificantly different (ANOSIM,=0.999,=0.001 for the NMDS and=0.9343,=0.001 for the PCoA analysis).

Fig.4 Plots of nonmetric multidimensional scaling (nMDS) ordination (A) and unweighted unifrac principal coordinates analysis (PCoA) (B).
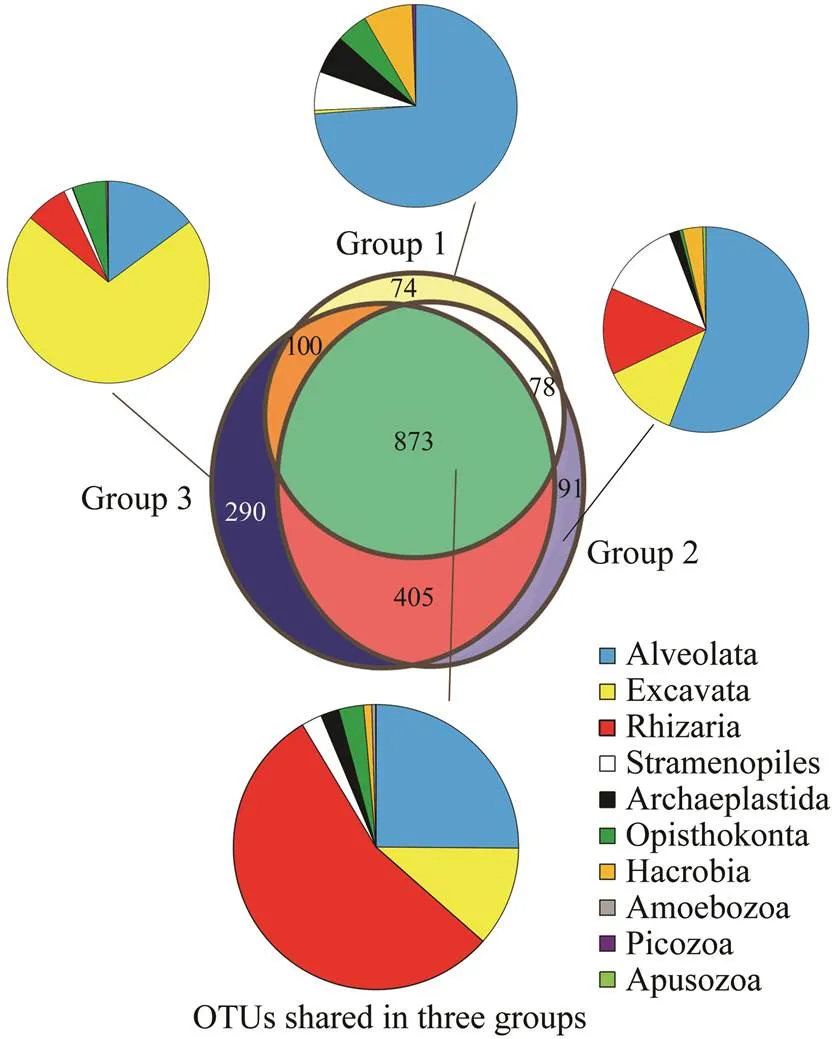
Fig.5 Venn diagram shows the OTUs shared among group 1 (samples G4.2000 and O14.4000), group 2 (samples H7.1000, O3.1000, and O14.1000), and group 3 (the rest samples) of protistan communities as indicated in Fig.4. Pie charts show the unique OTUs in each group and OTUs occurred in all three groups.
OTUs found only in group 1 were dominated by members in Alveolata (about 74%), followed by Hacrobia (about 14%). OTUs occurred only in group 2 were mainly composed of members in Alveolata (about 56%), followed by Rhizaria (about 14%) and Stramenopiles (13%). Group 3 specific OTUs were dominated by members in Excavata (about 71%), followed by Alveolata (about 15%). Approximately 45% of all OTUs were discovered from all three groups and were mainly composed of members in Rhizaria (55%), followed by members in Alveolata (25%) and Excavata (11%) (Fig.5).
3.2 Community Composition of Bathypelagic Protists
Pooling all data from all samples gave the first insight of the protistan community composition in the bathypelagic waters of the South China Sea. The reads affiliated with Rhizaria represented over half of the total protists, followed by Alveolata (26%) and Excavata (13%) (Fig.6A). The other supergroups, including Stramenopiles, Opisthokonta, Archaeplastida, Amoebozoa, Hacrobia and Picozoa, collectively contributed less than 8% to the total reads (Fig.6A). The community composition varied substantially in each sample,., Rhizaria dominated protistan communities in most samples except those retrieved from waters with 2000m depth at site G4 (G4.2000), 4000m depth at site O14 (O14.4000), and 1000m depth at site O3 (O3.1000) in which Alveolata replaced Rhizaria as the dominant taxon (Fig.6B). In at least half samples, Excavata surpassed Alveolata to be the second-most domi- nant group (Fig.6B). In terms of OTU richness, the domi- nant supergroup in the pooled dataset was Alveolata (about 47%), followed by Excavata (about 23%), Rhizaria (about 12%), and Stramenopiles (about 9%) (Fig.6C). This trend was consistent in all individual samples except G4.2000 and O14.4000 (Fig.6D).
The three dominant supergroups, includingRhizaria, Alveolata and Excavata, were further examined at lower taxonomic ranks (Fig.7). For Rhizaria, Spumellarida was the most dominant group (on average 85% of rhizarian reads) in all samples, followed by RAD-A (on average 10%) (Figs.7A, B). For Alveolata, Dinophyceae and MALV- I were the two most dominant groups, representing 35% and 27% of all reads, respectively. Members in MALV-II contributed 18% of Alveolata-affiliated reads. Apicom- plexa was the fourth most dominant supergroup; however, in samples O3.1000 and O3.2000, it replaced Dinophy- ceae to be the most dominant alveolate group with a con- tribution of 40% (Figs.7C,D). ForExcavata, members in Diplonemea and Neobodonid contributed equally and they constituted 91% of all Excavata-affiliated reads (Figs.7E,F). Phototrophic groups, including Bacillariophyta, Bolido- phyceae, Dictyochophyceae, Prasinophyceae, and Prymnesi- ophyceae, were retrieved from all samples and their con- tributions to total reads ranged from 0.3% to 16% in each sample (Figs.7G,H).
The ten most abundant OTUs in the pooled dataset belonged to Spumellarida (three OTUs), Discoba (three OTUs), RAD-A (two OTUs), and Dinophyceae (two OTUs). These OTUs had high similarities with environmental sequences in the GenBank database, and seven of them showed 100% similarities (Table 2). OTU_1 was the most abundant OTU and was a polycystine radiolarian that was 94.3% similar toIt was followed by OTU_3091 which showed 92.8% similarity to the same species whereas OTU_25 was identical to. OTU_10 and OTU_37 showed rather low similarities with their nearest named neighbor in the GenBank database, namelysp. (HQ651785) (79.4% and 80.3%, respectively). OTU_31 and OTU_35 were members of Discoba and showed 77.6% and 77.8% similarities with their nearest named neighbors(AB948221) andsp. (AF38 0997), respectively (Table 2). OTU_15 was also a member of Discoba and showed 100% similarity with(DQ207580). OTU_21 and OTU_74 showed high similarities with their nearest named neighbors,(AB686256) (100%) and thea toxic bloom-forming dinophyceaen,(JF79 1049) (99.2%).

Fig.6 Relative abundance of reads in pooled samples (A) and in each sample (B). Relative OTU richness in pooled samples (C) and in each sample (D).

Fig.7 Pie charts calculated for the percentage of total reads in all samples of different taxa from Rhizaria (A), Alveolata (C), Excavata (E), and phototrophic (G) groups. Histogram shows the relative abundances of reads from Rhizaria (B), Alveolata (D), Excavata (F), and phototrophic (H) groupsin each sample. Different groups are depicted in different colors.
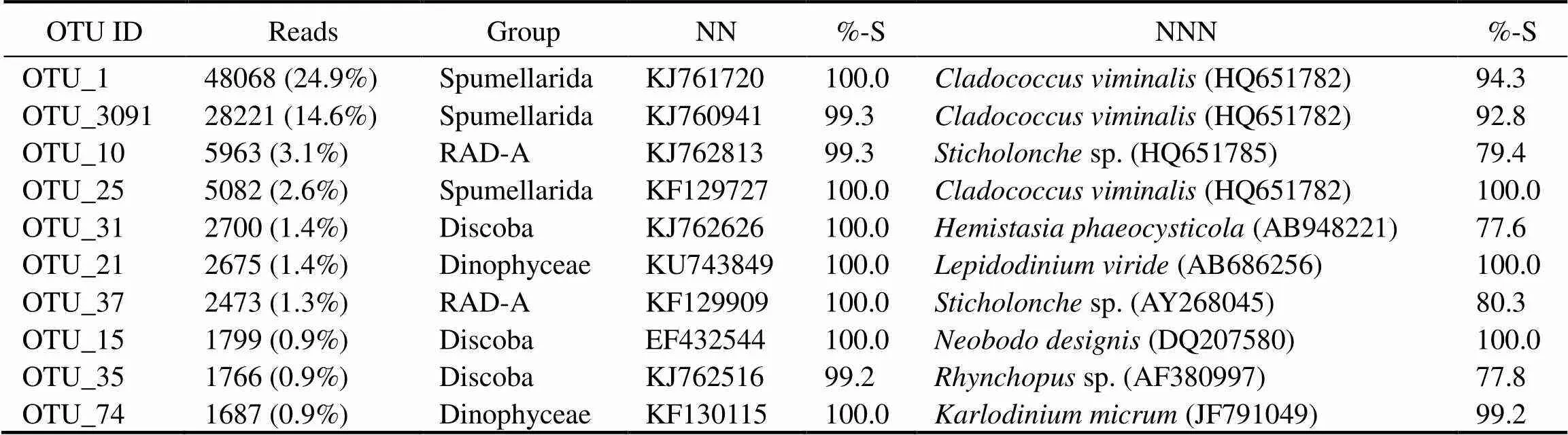
Table 2 List of the top ten most abundant OTUs
Notes: %-S, similarities; NN, the nearest neighbor; NNN, the nearest named neighbor.
4 Discussion
High-throughput sequencing, which can detect species at rather low abundance and bypass the difficulties of conventional morphology-based methods, was used to reveal the composition of protist communities in bathypelagic waters of the central South China Sea. The main aim was to characterize the community structure and diversity patterns of protists,which is helpful for understanding their roles in deep oceanic waters.
4.1 Protistan Community Compositionin the Bathyal Zone of the South China Sea
In the present study, the average alpha diversity was found to decrease as water depth increased, although no significant difference was found among sample groups. Previous studies have shown that bacterial richness declines by about 25% from the epipelagic layer to the bathypelagic waters (Moeseneder, 2001; Hewson, 2006) and temperature was proposed to be a major stratifying factor of bacterial communities overriding hydrostatic pressure (Martin-Cuadrado, 2007). Studies have also shown that protistan diversity declines with depth along the water columns (Countway, 2007; Xu, 2017b; Giner, 2019). The decline of protistan diversity with depth has been interpreted as an indication of a finite number of ecological niches present in the deep ocean for protists (Countway, 2007).
In terms of relative sequence abundances, members affiliated with Rhizaria constituted the most dominant group followed by Alveolata,though there were variabilities among individual samples. Excavata surpassed Stramenopiles to be the third most dominant group (Figs.5A,B). In terms of OTU richness, Alveolata was the most dominant group followed by Excavata and Rhizaria (Figs.5C,D). Rhizarians have been repeatedly identified to dominate deep sea protistan communities and, in some cases, they were also reported to possess the highest richness (Pernice, 2015; Xu, 2017a). In the present study, the dominant groups of rhizarians were RAD_A, B, C and Polycystinea, which is consistent with previous analyses of bathypelagic radiolarian communities (Pernice, 2015). A survey using clone library construction performed on the samples collected from the same cruise also showed the dominance of Rhizaria in deep waters with Spumellarida being the greatest contributor in this supergroup (Xu., 2017a). It is noteworthy that the cell-size of rhizarians is usually largerthan the majority of planktonic protists, the standing stock of which in the top 200m of the world’s oceans is estimated to be equivalent to 5.2% of the total oceanic biota carbon reservoir (Biard, 2016). Furthermore, rhizarians also contribute to the downward transportation of carbon via the so-called biological pump. For example, large colonial rhizarians of the order Collodaria have been shown to correlate significantly with downward fluxes of carbon (Guidi, 2016). Additionally, the large-celled members of Phaeodaria are reported to play a major role in carbon flux attenuation in the twilight zone in the California Current Ecosystem (Stukel., 2018). Another rhizarian group, Acantharia, has been shown to contribute to the particulate organic carbon fluxcyst formation down to meso-/bathy-pelagic waters in the Sou- thern Ocean and the Atlantic Ocean (Decelle, 2013; Belcher, 2018). Thus, the rhizarian-affiliated sequences retrieved from the deep sea were thought to derive from extracellular material of larger cells sinking from the upper waters (Not, 2009). Other data, however, do not support this conclusion. For example, novel rhizarian OTUs/clades have been reported, both in the deep oxygenated meso-/bathy-pelagic waters and in anoxic basins (Not., 2007a; Gilg., 2010; Orsi., 2011; Xu., 2017a). Furthermore, RNA-based sequence data, which is not subject to the same distortions as DNA-based sequence data (., sequences generated from the inactive/dead cells and extracellular nucleic acids), have confirmed the presence of active rhizarians in deep oceanic waters (Hu, 2016; Xu, 2017b). Radiolarians, which have characteristic cell struc- tures and photosynthetic symbionts, are classified as pho- tosynthetic organisms in surface waters. However, little knowledge has been known for their lifestyle in the bathy- pelagic zone where photosynthetic symbionts are not expected (Stoecker, 2009; Ishitani, 2012). Thus the unknown rhizarian species dwelling in the deep waters needs to be further studied. When combined with paired,., RNA- and DNA-based, molecular sequencing approaches, these data will serve as a basis to gain a better understanding of the ecological role of rhizarians and other protists in the deep sea.
It has been suggested that the universal primers targeting the SSU rRNA gene favor some eukaryotic clades over others, leading to bias in the resulting molecular datasets (Amaral-Zettler, 2009). Compared with the V4 region of the SSU rRNA gene, it has been suggested that the shorter V9 region might mitigate such biases (Pawlowski, 2011). This finding appears to be especially true for excavates and foraminiferans (Pawlowski, 2011). Recent studies have shown that diplonemids (Excavata, Diplonemea), a major component of Excavata, is the most diverse and the sixth most abundant eukaryotic taxon in the global plankton, with a huge undiscovered diversity in the deep ocean (de Vargas, 2015; Flegontova, 2016). In the present study, Excavata was found to be the third most abundant group in terms of relative sequence abundance, and the second most diverse group in terms of OTU richness in the deep waters of the South China Sea. Members in Diplonemea were the major contributors of Excavata which is consistent with previous reports (Figs.6E, F). Members of the family Neobodonidae (Excavata, Kinetoplastida) were also retrieved from the deep waters of the South China Sea and constituted abou8% of all sequences affiliated with Excavata (Figs.6E,F). In the deep dark ocean, protistan grazing is considered to be one of the major causes of prokaryote mortality, arguably surpassing viral lysis (Nagata, 2010). The deep sea protistan grazers are mostly small, nano-sized, flagellated cells.They are com- monly referred to as heterotrophic nanoflagellates (HNFs), which include a wide range of phylogenetically diverse microbial eukaryotes. Members in Neobododid are considered to be important protistan bacterivores in the ocean (Chavez-Dozal, 2013; Mukherjee, 2015). Using catalyzed reporter deposition fluorescence in situ hybridization (CARD-FISH), it has been reported that kinetoplastids represented a significant fraction (average 21.8%) of total eukaryotic microbes in the deep subtropical North Atlantic (Morgan-Smith, 2011, 2013). It has also been reported that neobodonids are dominant kinetoplastids in the global ocean (Flegontova, 2018). Thus neobodonids are probably a major component of deep sea HNF communities and may play key roles in deep sea biogeochemical cycling through active grazing on prokaryotes.
4.2 Phototrophic Protists in the Bathy Zone of the South China Sea
In recent years, signals from deep sea phototrophic cells (both prokaryotes and eukaryotes) have been repeatedly reported in studies using various approaches including microscopy, flow cytometry and DNA/RNA- based sequencing (Scharek, 1999; Jiao, 2014; Pernice, 2015; Xu, 2017b). For instance, viable diatoms were retrieved from the global deep ocean collected by customized bottle-net sampler followed by vital stain and DNA-based sequencing (Agusti, 2015). Using both SSU rRNA and p23S rRNA gene analysis, phototrophic signals were found in the Challenger Deep of the Mariana Trench (Guo, 2018). By integrating epifluorescence microscopy observation and sequencing of SSU rRNA and psbA gene transcripts, live/active pigmented nano-sized eukaryotes (primarily haptophytes) were detected in the deep Western Pacific Ocean (Xu, 2018). In the present study, the contribution of phototrophic signals (mainly from Bacillariophyta, Bolidophyceae, Dictyochophyceae, Prasinophyceae, and Prymnesiophyceae) to the total microbial eukaryote biota ranged from 0.3% to 16%, with an average value of 3% (Figs.6G,H). Previous studies have shown that phototrophic microbes make a significant contribution to the downward transportation of carbon via various fast-sinking mechanisms (Jiao, 2014; Cai, 2015; Pernice, 2015). Based on DNA rather than RNA sequencing, the present study further confirmed the presence of phototrophic microbial eukaryotes in the deep sea. It has also been proposed that deep sea phototrophic signals are possibly from mixotrophs as many planktonic protists have a mixotrophic mode of nutrition (Stoecker, 2017; Xu, 2018). In the present study, members of Prymnesiophyceae, some of which are mixotrophic organisms, were recovered. It indicates the possible existence of heterotrophy of these cells that can make themselves adapt to and survive in the bathypelagic environment.
Acknowledgements
We thank the captain and crew of thefor successful sampling. This work was supported by the National Natural Science Foundation of China (Nos. 41876142, 91751207 and 41861144018) and the Senior User Project of(No. KEXUE2019 G08) funded by the Center for Ocean Mega-Science, Chi- nese Academy of Sciences.
Adl, S. M., Bass, D., Lane, C. E., Lukeš, J., Schoch, C. L., Smirnov, A., Agatha, S., Berney, C., Brown, M. W., Burki, F., Cárdenas, P., Čepička, I., Chistyakova, L., Del Campo, J., Dunthorn, M., Edvardsen, B., Eglit, Y., Guillou, L., Hampl, V., Heiss, A. A., Hoppenrath, M., James, T. Y., Karnkowska, A., Karpov, S., Kim, E., Kolisko, M., Kudryavtsev, A., Lahr, D. J. G., Lara, E., Le Gall, L., Lynn, D. H., Mann, D. G., Massana, R., Mitchell, E. A. D., Morrow, C., Park, J. S., Pawlowski, J. W., Powell, M. J., Richter, D. J., Rueckert, S., Shadwick, L., Shimano, S., Spiegel, F. W., Torruella, G., Youssef, N., Zlatogursky, V., and Zhang, Q., 2019. Revisions to the classification, nomenclature, and diversity of eukaryotes., 66: 4-119.
Agusti, S., Gonzalez-Gordillo, J. I., Vaque, D., Estrada, M., Cerezo, M. I., Salazar, G., Gasol, J. M., and Duarte, C. M., 2015. Ubiquitous healthy diatoms in the deep sea confirm deep carbon injection by the biological pump., 6: 7608.
Amaral-Zettler, L. A., McCliment, E. A., Ducklow, H. W., and Huse, S. M., 2009. A method for studying protistan diversity using massively parallel sequencing of V9 hypervariable regions of small-subunit ribosomal RNA genes., 4: e6372.
Arístegui, J., Gasol, J. M., Duarte, C. M., and Herndl, G. J., 2009. Microbial oceanography of the dark ocean’s pelagic realm., 54: 1501-1529.
Azam, F., Fenchel, T., Field, J. G., Gray, J. S., Meyer-Rei, L. A., and Thingstad, F., 1983. The ecological role of water-column microbes in the sea., 10: 257- 263.
Bass, D., Moreira, D., López-García, P., Polet, S., Chao, E. E., von der Heyden, S., Pawlowski, J., and Cavalier-Smith, T., 2005. Polyubiquitin insertions and the phylogeny of Cercozoa and Rhizaria., 156: 149-161.
Belcher, A., Manno, C., Thorpe, S., and Tarling, G., 2018. Acantharian cysts: High flux occurrence in the bathypelagic zone of the Scotia Sea, Southern Ocean., 165: 117.
Biard, T., Stemmann, L., Picheral, M., Mayot, N., Vandromme, P., Hauss, H., Gorsky, G., Guidi, L., Kiko, R., and Not, F., 2016.imaging reveals the biomass of giant protists in the global ocean., 532: 504-507.
Bolger, A. M., Lohse, M., and Usadel, B., 2014. Trimmomatic: Aflexible trimmer for Illumina Sequence Data., 30: 2114-2120.
Cai, P., Zhao, D., Wang, L., Huang, B., and Dai, M., 2015. Role of particle stock and phytoplankton community structure in regulating particulate organic carbon export in a large marginal sea., 120: 2063-2095.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., Fierer, N., Peña, A. G., Goodrich, J. K., Gordon, J. I., Huttley, G. A., Kelley, S. T., Knights, D., Koenig, J. E., Ley, R. E., Lozupone, C. A., McDonald, D., Muegge, B. D., Pirrung, M., Reeder, J., Sevinsky, J. R., Turnbaugh, P. J., Walters, W. A., Widmann, J., Yatsunenko, T., Zaneveld, J., and Knight, R., 2010. QIIME allows analysis of high-throughput community sequencing data., 7: 335-336.
Caron, D. A., Alexander, H., Allen, A. E., Archibald, J. M., Armbrust, E. V., Bachy, C., Bharti, A., Bell, C. J., Dyhrman, S. T., Guida, S., Heidelberg, K. B., Kaye, J. Z., Metzner, J., Smith, S. R., and Worden, A. Z., 2017. Probing the evolution, ecology and physiology of marine protists using transcriptomics., 15: 6-20.
Caron, D. A., Countway, P. D., Jones, A. C., Kim, D. Y., and Schnetzer, A., 2012. Marine protistan diversity., 4: 467-493.
Caron, D. A., Worden, A. Z., Countway, P. D., Demir, E., and Heidelberg, K. B., 2009. Protists are microbes too: A perspective., 3: 4-12.
Chavez-Dozal, A., Gorman, C., Erken, M., Steinberg, P. D., McDougald, D., and Nishiguchi, M. K.,2013. From the laboratory into the field: Testing defense mechanisms of bacterial biofilms against protozoan grazing., 79: 553-558.
Countway, P. D., Gast, R. J., Dennett, M. R., Savai, P., Rose, J. M., and Caron, D. A., 2007. Distinct protistan assemblages characterize the euphotic zone and deep sea (2500m) of the western North Atlantic (Sargasso Sea and Gulf Stream)., 9: 1219-1232.
Decelle, J., Martin, P., Paborstava, K., Pond, D. W., Tarling, G., Mahé, F., de Varges, C., Lampitt, R., and Not, F., 2013. Diversity, ecology and biogeochemistry of cyst-forming Acantharia (Radiolaria) in the oceans., 8: e53598.
de Vargas, C., Audic, S., Henry, N., Decelle, J., Mahé, F., Logares, R., Lara, E., Berney, C., Le Bescot, N., Probert, I., Carmichael, M., Poulain, J., Romac, S., Colin, S., Aury, J. M., Bittner, L., Chaffron, S., Dunthorn, M., Engelen, S., Flegontova, O., Guidi, L., Horák, A., Jaillon, O., Lima-Mendez, G., Lukeš, J., Malviya, S., Morard, R., Mulot, M., Scalco, E., Siano, R., Vincent, F., Zingone, A., Dimier, C., Picheral, M., Searson, S., Kandels-Lewis, S., and Tara Oceans Coordinators, Acinas, S. G., Bork, P., Bowler, C., Gorsky, G., Grimsley, N., Hingamp, P., Iudicone, D., Not, F., Ogata, H., Pesant, S., Raes, J., Sieracki, M. E., Speich, S., Stemmann, L., Sunagawa, S., Weissenbach, J., Wincker, P., and Karsenti, E.,2015. Eukaryotic plankton diversity in the sunlit ocean.,348: 1261605.
Edgar, R. C., 2013. UPARSE: Highly accurate OTU sequences from microbial amplicon reads., 10: 996- 998.
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R., 2011. UCHIME improves sensitivity and speed of chimera detection., 27: 2194-2200.
Flegontova, O., Flegontov, P., Malviya, S., Audic, S., Wincker, P., de Vargas, C., Bowler, C., Lukeš, J., and Horák, A., 2016. Extreme diversity of diplonemid eukaryotes in the ocean., 26: 3060-3065.
Flegontova, O., Flegontov, P., Malviya, S., Poulain, J., de Vargas, C., Bowler, C., Lukeš, J., and Horák, A., 2018. Neobodonids are dominant kinetoplastids in the global ocean., 20: 878-889.
Gilg, I. C., Amaral-Zettler, L. A., Countway, P. D., Moorthi, S., Schnetzer, A., and Caron, D. A., 2010. Phylogenetic affiliations of mesopelagic Acantharia and acantharian-like environmental 18S rRNA genes off the southern California coast., 161: 197-211.
Giner, C. R., Balagué, V., Pernice, M. C., Duarte, C. M., Gasol, J. M.,Logares, R., and Massana, R., 2019. Marked changes in diversity and relative activity of picoeukaryotes with depth in the global ocean., DOI: https://doi.org/10.1101/552604.
Guidi, L., Chaffron, S., Bittner, L., Eveillard, D., Larhlimi, A., Roux, S., Darzi, Y., Audic, S., Berline, L., Brum, J., Coelho, L. P., Espinoza, J. C. I., Malviya, S., Sunagawa, S., Dimier, C., Kandels-Lewis, S., Picheral, M., Poulain, J., Searson, S., Tara Oceans coordinators, Stemmann, L., Not, F., Hingamp, P., Speich, S., Follows, M., Karp-Boss, L., Boss, E., Ogata, H., Pesant, S., Weissenbach, J., Wincker, P., Acinas, S. G., Bork, P., de Vargas, C., Iudicone, D., Sullivan, M. B., Raes, J., Kar- senti, E., Bowler, C., and Gorsky, G., 2016. Plankton networks driving carbon export in the oligotrophic ocean., 532: 465-470.
Guillou, L., Viprey, M., Chambouvet, A., Welsh, R. M., Kirkham, A. R., Massana, R., Scanlan, D. J., and Worden, A. Z., 2008. Widespread occurrence and genetic diversity of marine parasitoids belonging to Syndiniales (Alveolata)., 10: 3349-3365.
Guo, R., Liang, Y., Xin, Y., Wang, L., Mou, S., Cao, C., Xie, R., Zhang, C., Tian, J., and Zhang, Y., 2018. Insight into the pico- and nano-phytoplankton communities in the deepest biosphere, the Mariana Trench., 9: 2289.
Herndl, G. J., and Reinthaler, T., 2013. Microbial control of the dark end of the biological pump., 6: 718- 724.
Hewson, I., Steele, J. A., Capone, D. G., and Fuhrman, J. A., 2006. Remarkable heterogeneity in meso- and bathypelagic bacterioplankton assemblage composition., 51: 1274-1283.
Hu, S. K., Campbell, V., Connell, P., Gellene, A. G., Liu, Z., Terrado, R., and Caron, D. A., 2016. Protistan diversity and activity inferred from RNA and DNA at a coastal ocean site in the eastern North Pacific., 92: fiw050.
Hu, X., Lin, X., and Song, W., 2019.. Science Press, Beijing, 932pp.
Ishitani, Y., Ujiie, Y., de Vargas, C., Not, F., and Takahashi, K., 2012. Phylogenetic relationships and evolutionary patterns of the order Collodaria (Radiolaria)., 7: e35775.
Jiao, N., Herndl, G. J., Hansell, D. A., Benner, R., Kattner, G., Wilhelm, S. W., Kirchman, D. L., Weinbauer, M. G., Luo, T. W., Chen, F., and Azam, F., 2010.Microbial production of recalcitrant dissolved organic matter: Long-term carbon storage in the global ocean., 8: 593- 599.
Jiao, N., Luo, T., Zhang, R., Yan, W., Lin, Y., Johnson, Z. I., Tian, J., Yuan, D., Yang, Q., Sun, J., Hu, D., and Wang, P., 2014. Presence ofin the aphotic waters of the Western Pacific Ocean., 11: 2391-2400.
Li, R., Jiao, N., Warren, A., and Xu, D., 2018. Changes in community structure of active protistan assemblages from the lower Pearl River to coastal waters of the South China Sea., 63: 72-82.
Lozupone, C., and Knight, R., 2005. UniFrac: A new phylogenetic method for comparing microbial communities., 71: 8228-8235.
Magoč, T., and Salzberg, S. L., 2011. FLASH: Fast length adjustment of short reads to improve genome assemblies., 27: 2957-2963.
Martín-Cuadrado, A. B., López-García, P., Alba, J. C., Moreira, D., Monticelli, L., Strittmatter, A., Gottschalk, G., and Ro- dríguez-Valera, F., 2007. Metagenomics of the deep Mediterranean, a warm bathypelagic habitat., 2: e914.
Moeseneder, M. M., Winter, C., and Herndl, G. J., 2001. Horizontal and vertical complexity of attached and free-living bacteria of the eastern Mediterranean Sea, determined by 16S rDNA and 16S rRNA fingerprints., 46: 95-107.
Morgan-Smith, D., Herndl, G., van Aken, H., and Bochdansky, A., 2011. Abundance of eukaryotic microbes in the deep subtropical North Atlantic., 65: 103- 115.
Morgan-Smith, D., Clouse, M. A., Herndl, G. J., and Bochdansky, A., 2013. Diversity and distribution of microbial eukaryotes in the deep tropical and subtropical North Atlantic Ocean., 78: 58-69.
Mukherjee, I., Hodoki, Y., and Nakano, S., 2015. Kinetoplastid flagellates overlooked by universal primers dominate in the oxygenated hypolimnion of Lake Biwa, Japan.,91: fiv083.
Nagata, T., Tamburini, C., Arístegui, J., Baltar, F., Bochdansky, A. B., Fonda-Umani, S., Fukuda, H., Gogou, A., Hansell, D. A., Hansman, R. L., Herndl, G. J., Panagiotopoulos, C., Reinthaler, T., Sohrin, R., Verdugo, P., Yamada, N., Yamashita, Y., Yokokawa, T., and Bartlett, D. H., 2010. Emerging concepts on microbial processes in the bathypelagic ocean–Ecology, biogeochemistry, and genomics., 57: 1519-1536.
Not, F., del Campo, J., Balagué, V., de Vargas, C., and Massana, R., 2009. New insights into the diversity of marine picoeukaryotes., 4: e7143.
Not, F., Gausling, R., Azam, F., Heidelberg, J. F., and Worden, A. Z., 2007. Vertical distribution of picoeukaryotic diversity in the Sargasso Sea., 9: 1233-1252.
Orsi, W., Edgcomb, V., Jeon, S., Leslin, C., Bunge, J., Taylor, G. T., Varela, R., and Epstein, S., 2011. Protistan microbial observatory in the Cariaco Basin, Caribbean. II. Habitat specialization., 5:1357-1373.
Pawlowski, J., Christen, R., Lecroq, B., Bachar, D., Shahbazkia, H. R., Amaral-Zettler, L., and Guillou, L., 2011. Eukaryotic richness in the abyss: Insights from pyrotag sequencing., 6: e18169.
Pernice, M. C., Giner, C. R., Logares, R., Perera-Bel, J., Acinas, S. G., Duarte, C. M., Gasol, J. M., and Massana, R.,2015. Large variability of bathypelagic microbial eukaryotic communities across the world’s oceans., 10: 945-958.
Scharek, R., Tupas, L. M., and Karl, D. M., 1999. Diatom fluxes to the deep sea in the oligotrophic North Pacific gyre at station ALOHA., 182: 55-67.
Schlitzer, R., 2011. Ocean data view. http://odv.awi.de.
Song, W., Warren, A., and Hu, X., 2009.. Science Press, Beijing, 518pp.
Stoecker, D. K., Johnson, M. D., deVargas, C., and Not, F., 2009. Acquired phototrophy in aquatic protists., 57: 279-310.
Stoecker, D. K., Hansen, P. J., Caron, D. A., and Mitra, A., 2017. Mixotrophy in the marine plankton., 9: 311-335.
Stukel, M. R., Biard, T., Krause, J., and Ohman, M. D., 2018. Large Phaeodaria in the twilight zone: Their role in the carbon cycle., 63: 2579-2594.
Sun, P., Huang, L., Xu, D., Chen, N., and Warren, A., 2017. Marked seasonality and high spatial variation in estuarine ciliates are driven by exchanges between the ‘abundant’ and ‘intermediate’ biospheres., 7: 9494.
Xu, D., Jiao, N., Ren, R., and Warren, A., 2017a. Distribution and diversity of microbial eukaryotes in bathypelagic waters of the South China Sea., 64: 370-382.
Xu, D., Li, R., Hu, C., Sun, P., Jiao, N., and Warren, A., 2017b. Microbial eukaryote diversity and activity in the water column of the South China Sea based on DNA and RNA high through- put sequencing., 8: 1121.
Xu, D., Sun, P., Zhang, Y., Li, R., Huang, B., Jiao, N., Warren, A., and Wang, L., 2018. Pigmented microbial eukaryotes fuel the deep sea carbon pool in the tropical Western Pacific Ocean., 20: 3811-3824.
February 9, 2019;
April 28, 2019;
June 12, 2019
© Ocean University of China, Science Press and Springer-Verlag GmbH Germany 2019
. E-mail: dapengxu@xmu.edu.cn
(Edited by Qiu Yantao)
 Journal of Ocean University of China2020年1期
Journal of Ocean University of China2020年1期
- Journal of Ocean University of China的其它文章
- Circulation and Heat Flux along the Western Boundary of the North Pacific
- System Reliability Analysis of an Offshore Jacket Platform
- The Mineral Composition and Sources of the Fine-Grained Sediments from the 49.6˚E Hydrothermal Field at the SWIR
- Research Progress of Seafloor Pockmarks in Spatio-Temporal Distribution and Classification
- Application of the Static Headland-Bay Beach Concept to a Sandy Beach: A New Elliptical Model
- Climatology of Wind-Seas and Swells in the China Seas from Wave Hindcast