
Deoxyelephantopin induces ROS-mediated autophagy and apoptosis in human colorectal cancer in vitro and in vivo
2020-02-17 12:22ChimKeiChanKindLengTongPooiFongWongHabsahAbdulKadir
Chim-Kei Chan, Kind-Leng Tong, Pooi-Fong Wong✉, Habsah Abdul Kadir✉
1Biomolecular Research Group, Biochemistry Program, Institute of Biological Sciences, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia
2Department of Pharmacology, Faculty of Medicine, University of Malaya, 50603 Kuala Lumpur, Malaysia
ABSTRACT Objective: To systematically map the stepwise events leading to deoxyelephantopin-induced cell death of HCT116 human colorectal cancer cells and evaluate the effectiveness of deoxyelephantopin in vivo.Methods: HCT116 cells were treated with deoxyelephantopin at various concentrations and time points. Autophagy was confirmed by the detection of autophagosomes and autophagosomal proteins by electron microscopy and Western blotting assays, respectively,and then validated by siRNA knockdown. In addition, apoptosis was confirmed by the detection of apoptosis-related proteins.The intracellular reactive oxygen species (ROS) level was measured using flow cytometry. The growth inhibitory effect of deoxyelephantopin was further evaluated in vivo using a mouse xenograft model.Results: Deoxyelephantopin firstly elevated ROS production,which then triggered autophagic flux with the accumulation of autophagosomal proteins including LC3A/B, ATG5, and ATG7,followed by the induction of apoptosis via the intrinsic and extrinsic pathways. Pre-treatment with N-acetyl-L-cysteine, a ROS inhibitor, reversed both apoptosis and autophagy. The knockdown of LC3 prevented apoptosis induction which confirmed that deoxyelephantopin induced autophagy-dependent apoptosis in HCT116 cells. Accumulation of ROS also activated apoptosis via the mitogen-activated protein kinases signaling pathway. Furthermore,deoxyelephantopin also inhibited the PI3K/AKT/mTOR pathway,which then released the inhibition of autophagy. In vivo study further showed that deoxyelephantopin significantly suppressed the growth of HCT116 subcutaneous xenograft in nude mice.Conclusions: Our findings revealed that deoxyelephantopin elevates oxidative stress and induces ROS-dependent autophagy followed by apoptosis in HCT116 cells via the concerted modulation of multiple signaling pathways. These findings further support the development of deoxyelephantopin as a therapeutic agent for colorectal cancer.
KEYWORDS: Deoxyelephantopin; Autophagy; PI3K; mTOR;Colorectal cancer; MAPK
1. Introduction
Colorectal carcinoma (CRC) is one of the most dreaded malignancies and causes high mortality worldwide[1]. Current treatment options for colorectal cancer include adjuvant or neoadjuvant chemotherapy[2].However, many of the chemotherapeutic agents used are associated with serious adverse effects, inadequate efficacy, and resistance development, which necessitate the continuous search for more efficacious and safer anti-CRC drugs.
Targeting signaling pathways that modulate apoptosis and autophagy remains an attractive strategy to inhibit cancer cell growth. Apoptosis or programmed cell death type Ⅰ is mediated via the intrinsic and extrinsic pathways involving the mitochondria and death receptors,respectively. Reactive oxygen species (ROS) generating agents can modulate apoptosis via the activation of the mitogen-activated protein kinases (MAPK) family members, including stress-activated c-Jun NH2-terminal kinase (JNK), extracellular signal-regulated protein kinases (ERK1/2, p44/p42) and p38 MAPK[3]. Autophagy,a highly conserved cellular degradation process that eliminates aged/malfunctioning organelles or misfolded proteins via the interaction between autophagosomes and lysosomes, also plays an important role in determining tumor fate[4]. Autophagy can be either cytoprotective or cytodestructive. Exploiting autophagy with its pro-death effect will enhance the efficacy of chemotherapeutic agents[5]. Compounds such as rottlerin and betulinic acid have been reported to induce both autophagy and apoptosis in prostate cancer stem cells and HT29 cells, respectively[6,7]. In addition,autophagy and apoptosis also share common signaling pathways such as phosphoinositide-3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) and MAPK[3,8,9].Therefore, a drug that simultaneously targets multiple survival pathways could increase its efficacy in inhibiting tumor growth and restore the sensitivity of the tumor cells to its cytotoxic effects.
To date, there remains strong interests to discover naturally-derived anti-cancer agents that target multiple signaling pathways important for tumor growth and progression, as alternatives for safer and more efficacious drugs[10,11]. Deoxyelephantopin (DET) is one of the major bioactive compounds isolated from Elephantopus scaber L., a popular Ayurvedic herb employed for the treatment of cancer,hepatitis, bronchitis, pneumonia and fever[12,13]. Previous studies have demonstrated the potentials of utilizing DET to target breast,cervical, nasopharyngeal, lung and liver cancer cells via various cell death mechanisms including apoptosis, cell cycle arrest, autophagy,paraptosis involving oxidative stress, nuclear factor-kappa B (NF-κB),PI3K/AKT, signal transducer and activator of transcription (STAT) and MAPK signaling pathways[14-19]. Our previous findings revealed that DET is a highly potent cytotoxic agent (IC50= 0.73 μg/mL) against HCT116 cells[20,21]in comparison to normal colon cells. Thus far,it is known that DET could induce HCT116 cell death via apoptosis and/or autophagy[17]but the stepwise events leading to cell death and the exact interplay between autophagy and apoptosis remains to be defined further. Moreover, the effect of DET has not been validated in vivo for CRC. Here, we used HCT116 cells as a model to systematically map the stepwise events leading to cell death and to investigate the anti-cancer effect of DET in vivo and identify the mechanisms involved.
2. Materials and methods
2.1. Reagents and antibodies
DET (purity ≥ 98%) was isolated from Elephantopus scaber L. and identified using spectroscopic analysis (1H,13C NMR and HRESIMS) as reported previously[20]. Roswell Park Memorial Institute(RPMI 1640) medium, dimethylsulfoxide (DMSO), N-acetyl-Lcysteine (NAC), 2'-7'-dichlorofluorescein diacetate (DCFH-DA)dye, acridine orange (AO), bovine serum albumin, fetal bovine serum and phosphate-buffered saline (PBS) were obtained from Sigma Aldrich (St Louis, MO, USA). Antibiotic-antimycotic and TrypLE were purchased from Thermo Fisher Scientific (Carlsbad,CA, USA). MITO-ID®membrane potential detection kit was purchased from Enzo Life Sciences (Lausen, Switzerland). Primary antibodies and anti-mouse/rabbit immunoglobulin G-horseradish peroxidase-conjugated secondary antibodies (Supplementary Table) for Western blotting and immunohistochemical analyses were purchased from Cell Signaling Technology (Danvers, MA,USA) and Thermo Fisher Scientific (Massachusetts, USA). Clarity enhanced chemiluminescence detection kit was purchased from Bio-Rad Laboratories (Hercules, CA, USA). RayBio®Human Apoptosis Antibody Array kit was purchased from RayBiotech Life (Norcross,GA, USA). The LC3 small interfering RNA (siRNAs) and scrambled siRNA were obtained from OriGene Technologies, Inc. (Rockville,Maryland, USA). DharmaFECT transfection reagents were purchased from GE Healthcare Life Sciences (Massachusetts, USA).
2.2. Cell culture
HCT116 cells were obtained from the American Type Culture Collection (Manassas, VA, USA). All cells were cultured in RPMI 1640 medium supplemented with 10% v/v heat-inactivated fetal bovine serum, and 0.01% antimycotic solution. The cells were maintained at 37 ℃ in a humidified atmosphere with 5% CO2.
2.3. Protein array analysis
HCT116 cells (1 × 106cells) were treated with DET (1.5 and 3.0 μg/mL) for 24 h in 60 mm2culture dishes. Negative control cells were treated with vehicle DMSO. Then, the cells were harvested and resuspended in lysis buffer containing protease and phosphatase inhibitors. Protein lysates were analyzed using human apoptosis antibody array (RayBiotech Life; Norcross, GA, USA) according to the manufacturer's protocol. Fluorescence intensities were measured by densitometry and fold change was determined in comparison to controls.
2.4. Detection of mitochondrial membrane potential
Mitochondrial dysfunction is often associated with changes in mitochondrial membrane potential which can be examined by using the cell-permeable fluorescent cationic dye, mito-ID. HCT116 cells(1 × 106cells) were cultured in 60 mm2culture for 24 h followed by treatment with different concentrations (0.75-3.0 μg/mL) of DET or vehicle DMSO only (untreated control) for 24 h. Subsequently, the cells were harvested and washed. The cell pellet was resuspended with culture media containing mito-ID and incubated at 37 ℃ in 5% CO2incubator for 15 min. Then, cell suspension was washed and replaced with growth medium. The detection of green and red fluorescence signals in FL1-A and FL2-A channels was performed using flow cytometry.
2.5. Detection of acidic vesicular organelles
To detect acidic vesicular organelle, cells were stained with AO and visualized by fluorescence microscopy. HCT116 cells (1 × 106cells) were treated with or without DET in the presence or absence of NAC for 24 h. After treatment, the cells were harvested, washed and incubated with 1 μg/mL of AO for 15 min at 37 ℃ and then washed again with PBS. The cells were viewed under a fluorescent microscope.
2.6. Transmission electron microscopy (TEM)
To elucidate the formation of autophagosomes mediated by DET in HCT116 cells, ultrastructural alterations were visualized by TEM.The treated cells were harvested and fixed with 4% glutaraldeyde and further fixed with 1% osmium tetroxide, which was followed by a series of dehydration process in increasing concentrations of alcohol. The cells were then further embedded in epon resin.Ultrathin sections were obtained by using an ultramicrotome(Leica Ultracut, Germany). The sections were placed onto copper grids and stained with uranyl acetate replacement and lead citrate.Subsequently, the sections were analyzed by Carl Zeiss Libra 120 energy filtering transmission electron microscope (Oberkochen,Germany).
2.7. Western blotting analysis
HCT116 cells (1 × 106cells) were cultured in 60 mm2culture dishes and treated with 3.0 μg/mL of DET at different durations of incubation (6, 12 and 24 h). Negative control cells were treated with vehicle DMSO at different durations of incubation (6, 12 and 24 h).The cells were then harvested, washed with cold PBS and incubated with cold radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitors on ice for 5 min. Cell lysates were obtained by centrifugation at 14 000 × g for 15 min at 4 ℃. Bradford assay was used to estimate the total content of protein. Twentyfive micrograms of the total protein of each lysates were loaded and separated by electrophoresis on a 10 or 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis gel. The proteins were transferred from gel onto 0.45 μM nitrocellulose membrane and the membrane was blocked by using skim milk or bovine serum albumin for 1 h.Next, the membranes were incubated with primary antibodies at 4 ℃overnight and then washed with 0.05% Tween 20 in tris-buffered saline(TBST). Next, the membrane was incubated with corresponding antimouse/rabbit immunoglobulin G-horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature and washed thrice with TBST. Finally, the membrane was incubated with enhanced chemiluminescence reagent and visualized using a gel documentation system. The protein band intensities were quantitated using Vilber Lourmart software.
2.8. Treatment with apoptosis and autophagy inhibitors
To validate the role of apoptosis and autophagy in DET-induced cell death, apoptosis inhibitors, carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (z-VAD-FMK) and autophagy inhibitors, 3-methyladenine (3-MA) were used to assess the effects of the inhibition of apoptosis and autophagy on DET-treated HCT116 cells. Briefly, HCT116 cells were pre-treated with 20 μM of z-VAD-FMK or 2.5 mM of 3-MA for 1 h prior to treatment with DET (3.0 μg/mL) for 24 h. Total proteins of the treated cells were then harvested for Western blotting analysis.
2.9. RNA interference of LC3
HCT116 cells were seeded 24 h prior to transfection. A final concentration of 25 nM of siRNA LC3 (siLC3) or scrambled negative control siRNA was transfected into the cells by using DharmaFECT transfection reagent following the manufacturer's protocol. The knockdown efficiency of 3 unique siRNAs (siRNA A, B and C)was measured by the abundance of protein using Western blotting analysis. siRNA with the best knockdown efficiency was used for subsequent experiments. For treatment, the transfected cells were treated with to 3.0 μg/mL of DET for an additional 24 h and then harvested for subsequent experiments.
2.10. Assessment of intracellular ROS level
To investigate the association between oxidative stress and cell death mechanisms, accumulation of intracellular ROS was examined by using DCFH-DA. HCT116 cells (1 × 106cells) were plated onto 60 mm2culture dish prior to DET treatment. DMSO was used as vehicle control while tert-butyl hydroperoxide was used as positive control. After incubation with 0.75, 1.5 and 3.0 μg/mL of DET for 4 h, the cells were washed and stained with 50 μM DCFH-DA in culture media. After an hour of incubation, cells were harvested and washed with PBS. PBS was then added to resuspend the cell pellet and the fluorescence intensity was measured by flow cytometry and detected in FL1-A channel. The effects of ROS inhibition on DETinduced autophagy and apoptosis in HCT116 cells were assessed by pre-treatment with NAC, a ROS inhibitor. Briefly, 1 mM of NAC was pre-incubated in HCT116 cells for 1 h prior to treatment with DET (3.0 μg/mL) for 24 h. Total proteins of the treated cells were then harvested for Western blotting analysis.
2.11. Treatment with p38, JNK and ERK specific inhibitors
To assess the involvement of MAPK pathway in DET-induced apoptosis, p38 specific inhibitor (SB202190), JNK specific inhibitor(SP600125) and ERK specific inhibitor (UO126) were used to inhibit the activation of this pathway prior to DET treatment. Briefly,HCT116 cells were pre-incubated with 10 μM of SB202190 or 5 μM of SP600125 or 5 μM of UO126, respectively, for 1 h prior to treatment with DET (3.0 μg/mL) for 24 h. Total proteins of the treated cells were then harvested for Western blotting analysis.
2.12. Ethics statement
Experiment with mice was performed in accordance to the protocol approved by the Faculty of Medicine Institutional Animal Care and Use Committee, University of Malaya (Ethics Reference Number:2014-03-05/IBS/R/CCK). Guidelines for the welfare and use of animals for cancer research were referred[22]. The entire experiment was performed in the AAALAC International accredited Animal Experimental Unit of the Faculty of Medicine, University of Malaya.
2.13. Human CRC xenograft experiment
Twenty-four female (BALB/c-nu) 4-week-old nude mice (15-20 grams) were purchased from InVivos Pte. Ltd. (Singapore) and housed in individually ventilated cages in the Animal Experimental Unit at University Malaya under pathogen-free conditions and supplied with sterilized food and water ad libitum. On day 0,HCT116 human CRC cells (1 × 106cells) with 50% matrigel (0.1 mL; Becton Dickinson, US) were inoculated subcutaneously into the right flank of each nude mouse. Once the tumors were palpable(within the range of 100 mm3), nude mice were randomly assigned into four groups with six nude mice in each group. Baseline tumor size and body weight were recorded prior to the treatment. All mice groups received intraperitoneal injection of 25 mL/kg of saline with 4% DMSO, 1.25 mg/kg DET, 2.5 mg/kg DET or 50 mg/kg 5-fluorouracil thrice a week for 28 d. Treatment of nude mice with 5-fluorouracil served as positive control group. The changes of body weight and tumor growth of each mouse were monitored and measured once a week over 4 weeks. The tumor volume was calculated using the following formula, 0.5 × length × width2. The mice were sacrificed once the tumor nodules have reached 1.5 cm in diameter. Tumors were fixed in 10% buffered formalin and embedded in paraffin wax.
2.14. Immunohistochemical (IHC) analysis
For IHC staining, paraffin-embedded tumors were sectioned into 4 μm thick and were deparaffinized in xylene and hydrated with graded alcohol, and incubated with 0.3% hydrogen peroxide (H2O2)for 10 min. Antigen unmasking was carried out by incubating slides in boiled 10 mM sodium citrate buffer (pH 6.0) for 30 min. The slides were then blocked with 5% normal goat serum for 1 h at room temperature and followed by incubation with antibodies overnight at 4 ℃. Subsequently, the slides were washed three times with wash buffer (1 × TBST) and incubated with secondary antibody for 30 min at room temperature. The slides were washed with TBST for three times for 5 min and exposed to 3, 3'-diaminobenzidine chromogen and counterstained in hematoxylin. For quantification, three random× 400 microscopic fields per slide were captured using an inverted microscope Nikon Eclipse TS100 (Nikon, Japan) and analyzed using Nikon NIS-BR element software (Nikon, Japan).
2.15. Statistical analysis
All assays were performed in at least three separate experiments and the data were stated as mean ± standard error (SE). Statistical analyses were performed using one-way analysis of variance(ANOVA) followed by Dunnett's test or Student's t-test, where applicable. P value < 0.05 was regarded statistically significant.
3. Results
3.1. Intrinsic and extrinsic apoptosis in HCT116 cancer cells induced by DET treatment
Protein array results revealed significant dose-dependent upregulation of p53, p21, caspase-3, FAS, death receptor 4 (DR4),DR5 and insulin-like growth factor-binding protein 5 (IGFBP-5)expressions and significant downregulation of X-linked inhibitor of apoptosis protein (XIAP), Survivin and insulin-like growth factor-1 receptor (IGF-1R) expressions (Figure 1A and B). Consistent with the protein array results, Western blotting analysis demonstrated time-dependent upregulation of DR5 and FAS expressions concomitant with decreased expressions of procaspase-9, -8 and-10 in HCT116 cells following treatment with DET (Figure 1C). In addition, inhibitor of apoptosis proteins (IAPs) such as XIAP and Survivin were downregulated in HCT116 cells after 24 hours of DET treatment (Figure 1C). Treatment with DET also increased the pro-apoptotic B-cell lymphoma 2 antagonist/killer (BAK) and Bcl-2-like protein 4 (BAX) expressions and markedly downregulated the expressions of anti-apoptotic B-cell lymphoma 2 (Bcl-2) compared to the vehicle control (Figure 1C). Disruption of mitochondria membrane potential was measured by mito-ID membrane potential fluorescent dye. Results showed prominent elevation of green monomer fluorescence intensity in the cytoplasm accompanied by the almost complete loss of orange aggregate fluorescence with increasing concentrations of DET-treated cells compared to the abundant orange aggregates in untreated cells (Figure 1D). The dosedependent reduction of mitochondrial membrane potential by DET thus suggests the involvement of mitochondria-mediated apoptosis in HCT116 cells. Collectively, this data confirmed that DET induces apoptosis via both the intrinsic and extrinsic pathways.
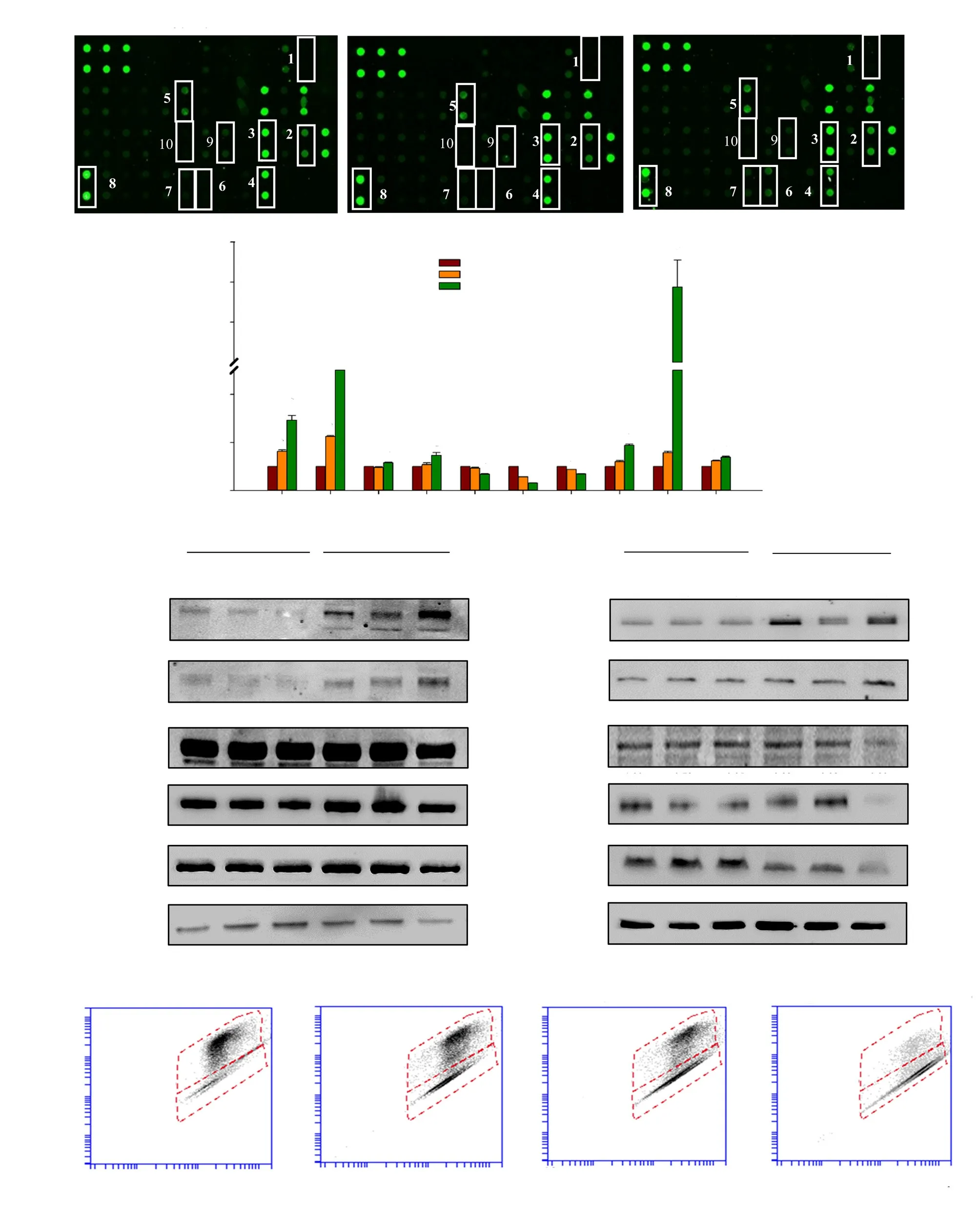
Figure 1. Apoptosis via the intrinsic and extrinsic pathways induced by deoxyelephantopin (DET). (A) Representative images of protein array. (B) Fold change of apoptosis-related proteins after DET treatment. 1- Caspase-3, 2- p53, 3- p21, 4- XIAP, 5- FAS, 6- DR4, 7- DR5, 8- Survivin, 9- IGF-1R and 10- IGFBP-5. (C)Apoptosis-related protein expressions of DET-treated cells at 6-24 h. The numbers above each band represent mean fold change of three separate experiments.(D) Mitochondrial membrane potential detected by mito-ID staining. Data are expressed as mean ± SE from three separate experiments. *P<0.05 compared to the control.
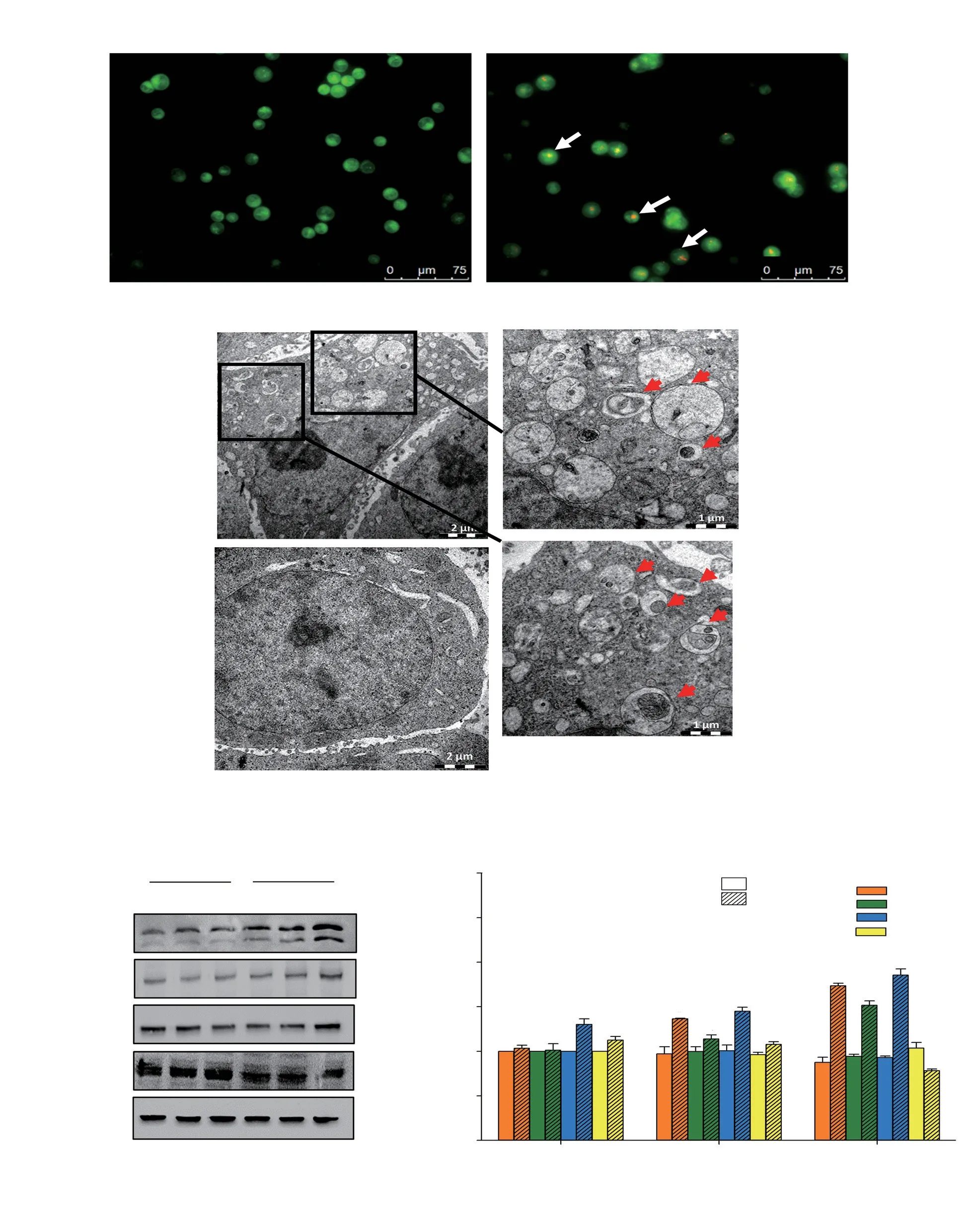
Figure 2. Autophagy in HCT116 cells after deoxyelephantopin (DET) treatment. (A) Acidic vesicular organelle induction (white arrows indicating positive cells) following treatment with DET. Magnification = 200×. (B) Ultrastructure of autophagosomes in HCT116 colorectal cancer cells treated with or without DET for 24 h. Red arrows indicate autophagosomes with residual digested materials. Bars = 2.0 μm and magnification = 1 200×(left). Bars = 1.0 μm and magnification = 3 200×(right). Protein expression levels of autophagy proteins are presented in (C) representative blots and (D) bar chart. GAPDH was used as loading control. Data are expressed as mean ± SE from three separate experiments. *P<0.05 compared to untreated control.
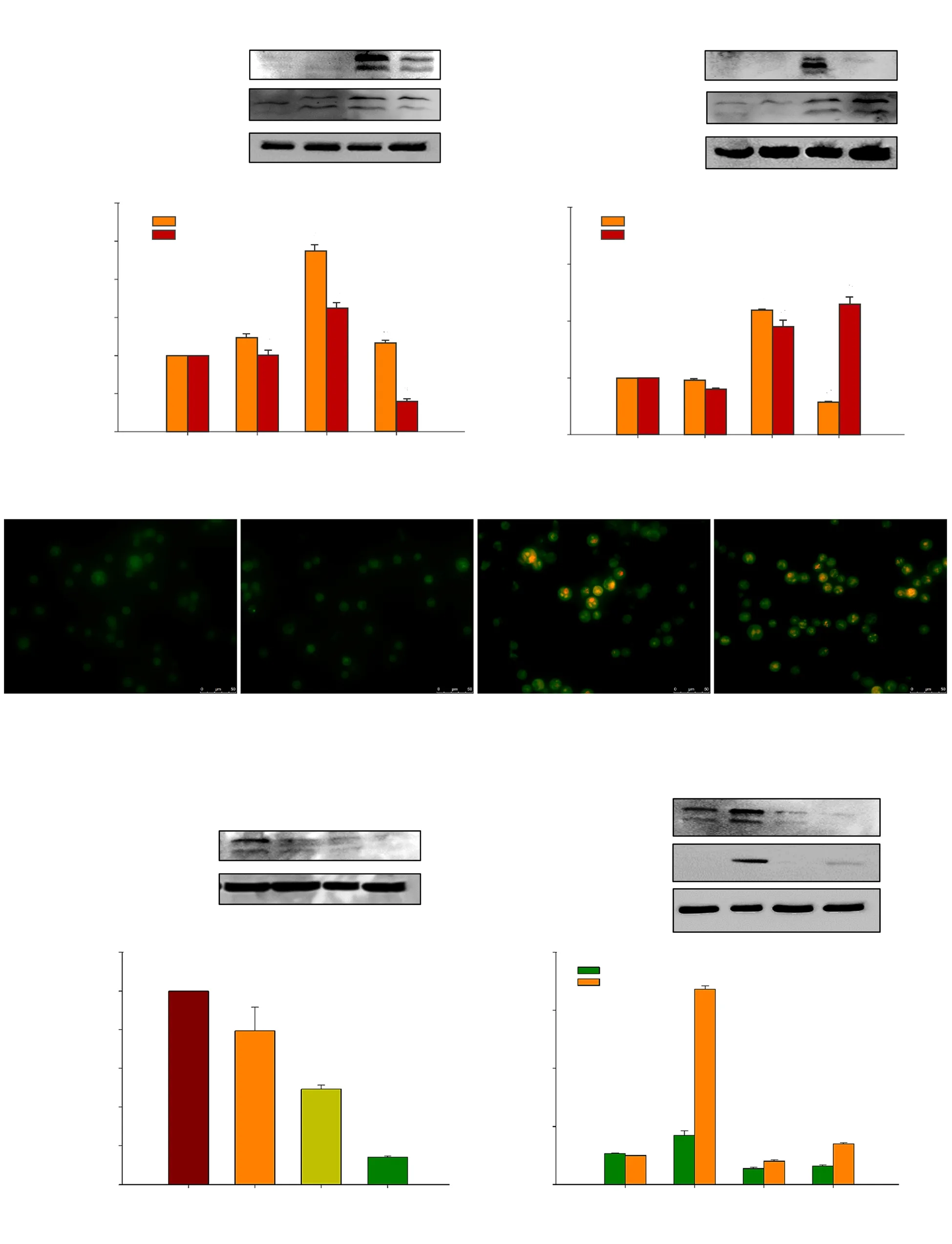
Figure 3. Autophagy-dependent apoptosis in HCT116 cells induced by deoxyelephantopin (DET). Cleaved caspase-3 and LC3A/B expressions in cells treated with 3-MA (autophagy inhibitor) (A, B) or z-VAD-FMK (apoptosis inhibitor) (C, D) before DET treatment. Acidic vesicular organelles in DET-treated cells,with or without z-VAD-FMK (E). Magnifciation = 400×. Relative protein expression after knockdown with three different siLC3 (F, G). LC3 and cleaved PARP expressions in siLC3-knockdown cells (H, I). GAPDH and β-actin were used as loading controls. Data are expressed as mean ± SE of three separate experiments. *P<0.05, #P<0.05 compared to the control and DET treatment group, respectively.
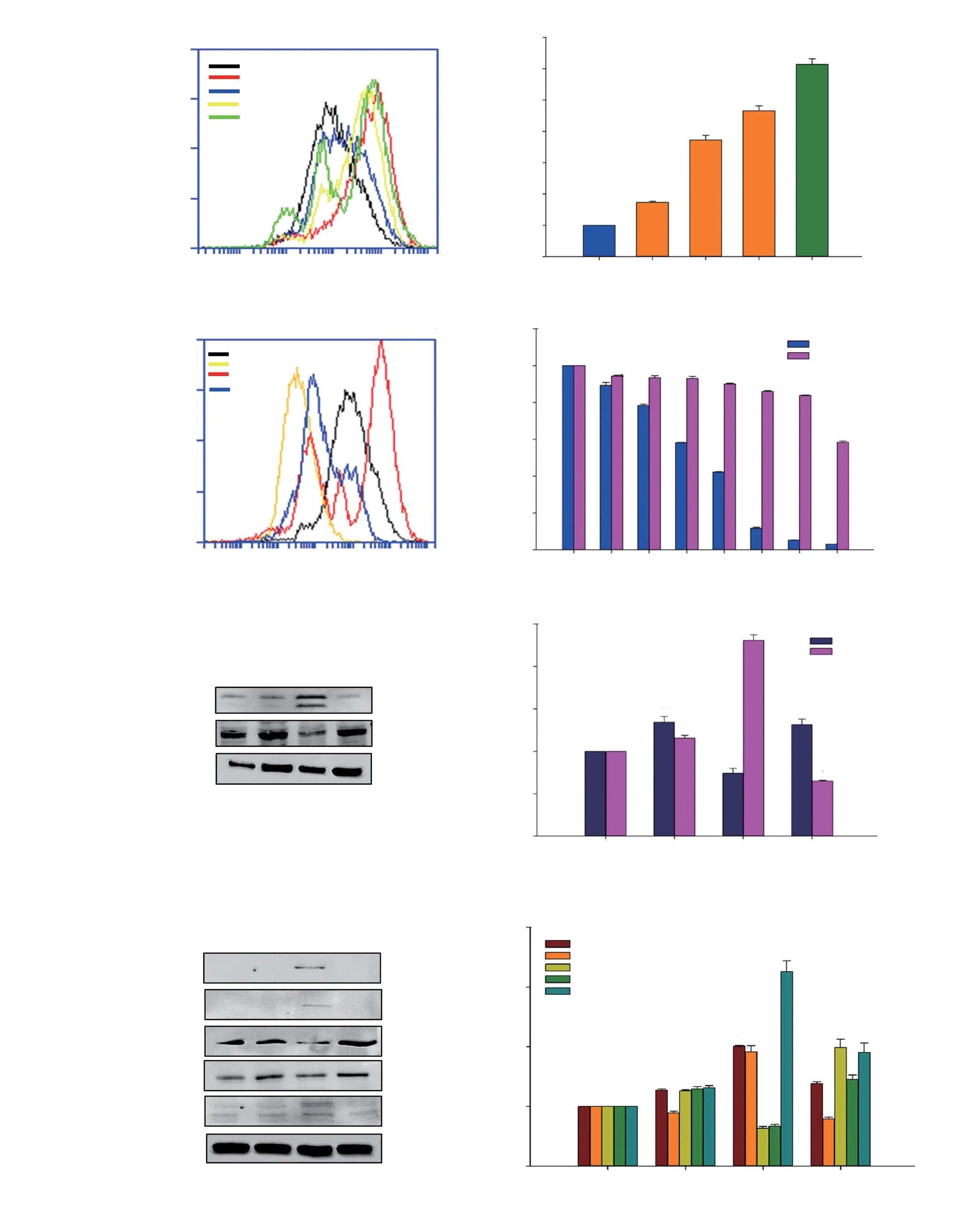
Figure 4. Oxidative stress in HCT116 cells induced by DET. Histogram (A) and bar chart (B) show intracellular ROS generation. Tert-butyl hydroperoxide(TBHP) served as positive control. ROS level (C), cell cytotoxicity (D), representative blots (E) and bar chart (F) of LC3A/B and p62 expressions,representative blots (G) and bar chart (H) of apoptosis-related protein expressions of DET-treated cells after NAC (ROS inhibitor) treatment. Data are expressed as mean ± SE of three separate experiments. *P<0.05, **P <0.05, #P <0.05 compared to the control, non-NAC treated group and DET-treated group,respectively.
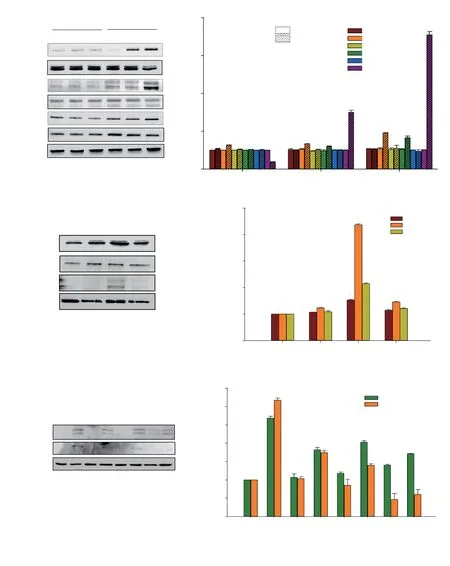
Figure 5. Involvement of ROS and the MAPK pathway in DET-induced apoptosis in HCT116 cells. Representative blots (A) and bar chart (B) of MAPK protein expressions in DET-treated cells at 6-24 h. Representative blots (C) and bar chart (D) of MAPK protein expressions in DET-treated cells after NAC treatment. Representative blots (E) and bar chart (F) of the expression of apoptosis-related proteins after MAPK inhibitors treatment. GAPDH was used as loading control. Data are expressed as mean ± SE from three separate experiments. *P<0.05 compared to the control. #P<0.05 compared to DET-treated group.
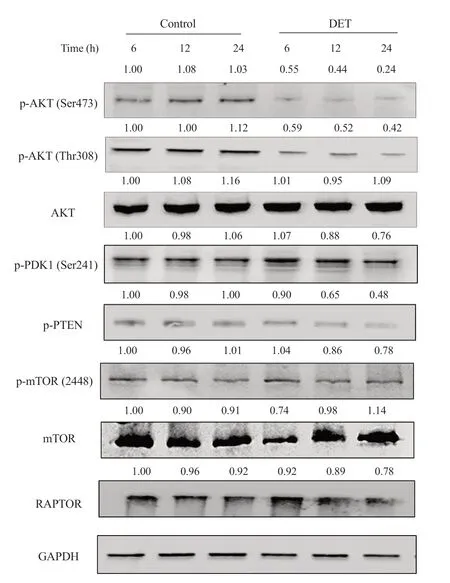
Figure 6. The PI3K/AKT/mTOR signaling pathway after DET treatment. HCT116 cells were treated with DET at different durations of incubation (6, 12 and 24 h). A representative Western blot of protein bands of AKT, phosphorylated (p)-AKT (Ser473), p-AKT (Thr308), p-PDK1, p-PTEN, p-mTOR, mTOR and RAPTOR is presented. GAPDH was used as loading control. The numbers above each band represent their respective mean fold change collectively from three separate experiments.
3.2. DET induced autophagy in HCT116 cancer cells
Treatment with DET resulted in the formation of reddish orange acidic vesicles resembling autolysosomes in the treated cells (Figure 2A). To further verify the occurrence of autophagy, cytological changes in DET-treated cells were examined by TEM. TEM images revealed the presence of numerous autophagosomes, which were doublemembrane vesicles containing residual digested materials in the DETtreated cells, whereas autophagosomes were absent in the control cells(Figure 2B). In addition, the results of Western blotting assays showed accumulations of autophagy-related proteins LC3A/B-Ⅱ, ATG7 and ATG5 in a time-dependent manner (Figure 2C and D). Conversely,DET reduced p62 protein expression in HCT116 cells after 24 hours of treatment.
3.3. Inhibition of autophagy induced apoptosis while the inhibition of apoptosis promoted autophagy in DET-treated cells
Co-treatment with 3-MA reduced the expression of LC3A/B-Ⅱ,indicating that the induction of autophagy was mitigated (Figure 3A and B). The suppression of autophagy in turn reduced the induction of apoptosis by DET to some extent as evidenced by moderate reduction of cleaved caspase-3 expression. As expected, pre-incubation with apoptosis inhibitor, z-VAD-FMK significantly inhibited the cleavage of caspase-3 in DET-treated cells (Figure 3C and D). Furthermore, z-VADFMK markedly enhanced the accumulation of LC3A/B-Ⅱ proteins,which was further supported by the significant accumulation of bright red acidic vesicles upon exposure of z-VAD-FMK inhibitor prior to DET treatment compared to DET treatment alone (Figure 3E). These results suggested that inhibition of autophagy induces apoptosis while inhibition of apoptosis promotes autophagy.
3.4. Inhibition of apoptosis via knockdown of LC3
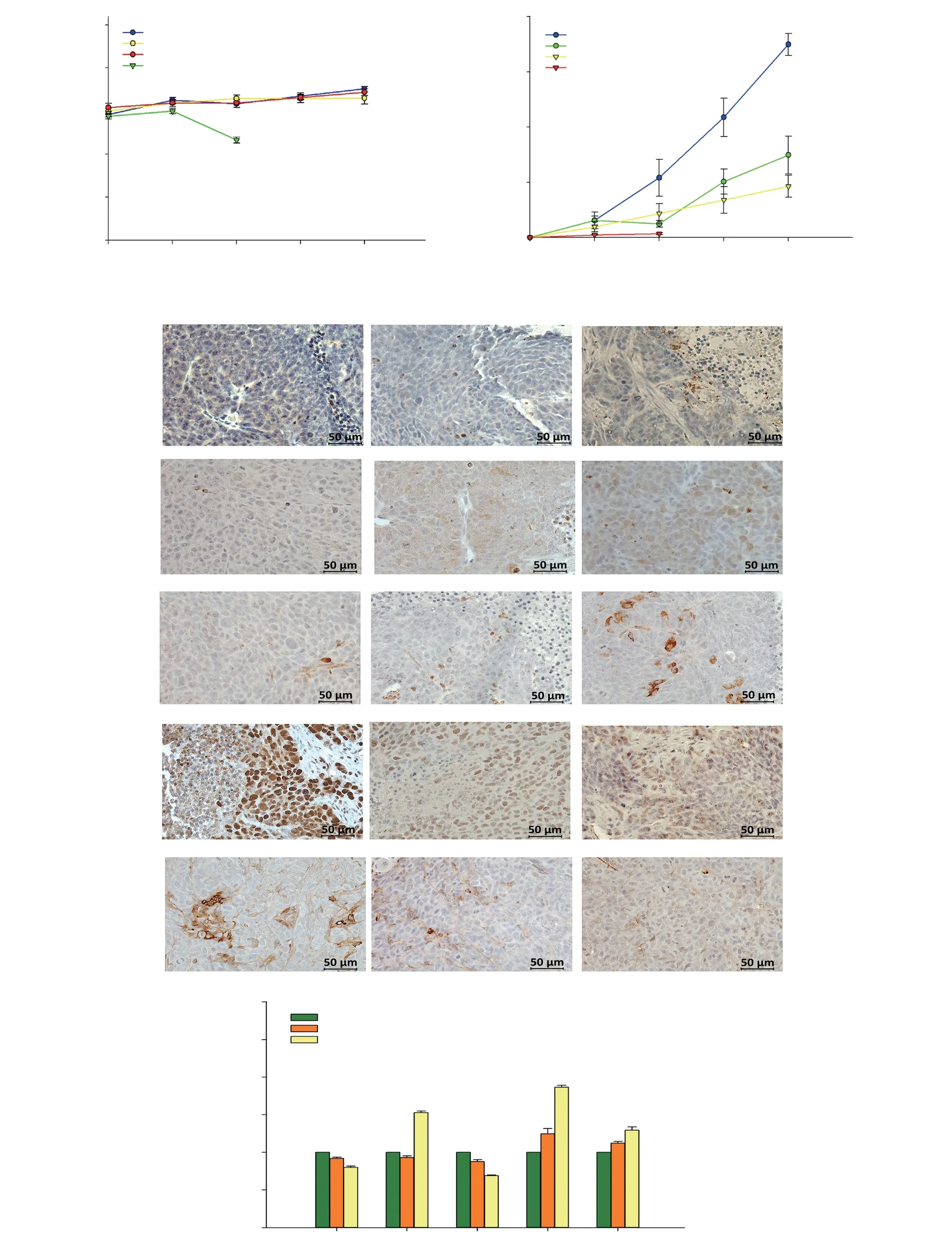
Figure 7. Anti-tumor effect of DET in HCT116 xenograft mouse model. Body weight (A) and tumor volume (B) of DET- and 5-fluorouracil (5-FU)-treated nude mice measured once a week over four weeks. 5-FU served as positive drug control. (C) Immunohistochemical images of apoptosis- and autophagyrelated protein expressions (positive cells are stained brown). Bars = 50 μm and magnification = 400×. (D) Fold change of positively stained cells of immunohistochemical images of DET-treated mice compared to vehicle controls. Data are expressed as mean ± SE (n=6). *P<0.05 compared to the control.
The pro-death effect of autophagy induced by DET was further confirmed by silencing LC3, which facilitated the formation of autophagosomes. The protein expression of LC3 was firstly assessed by Western blotting analysis to determine the knockdown efficiency of three unique siRNA duplexes targeting LC3 (siRNA A, B and C). Among the three siRNA duplexes, siRNA C gave the highest knockdown efficiency, with ~85.9% protein expression suppression(Figure 3F and G) with viability >100% (Supplementary Figure 1)in HCT116 cells and hence, siRNA C was chosen for subsequent experiments. Accumulation of LC3A/B-Ⅱ was significantly reduced after transfection with siRNA LC3 prior to the treatment with DET(Figure 3H). Knockdown of LC3 expression partially reversed DETinduced cytotoxicity in HCT116 cells (Supplementary Figure 1).Furthermore, the expression of apoptosis marker, cleaved PARP was reduced, indicating the apoptotic effect triggered by DET was interrupted in the absence of autophagy (Figure 3H and I).These findings confirmed that DET activates autophagy-dependent apoptosis in HCT116 cells.
3.5. Oxidative stress and ROS-dependent autophagy followed by apoptosis in HCT116 cells upon DET treatment
Upon exposure to increasing concentrations of DET (0.75, 1.5 and 3.0 μg/mL), the intracellular ROS level was significantly elevated (Figure 4A and B). The production of ROS was completely abolished by pre-treatment with NAC, a ROS inhibitor in the DETtreated cells (Figure 4C). In addition, the percentage of viable DETtreated cells also increased in the presence of NAC when compared to those without NAC (Figure 4D). This result confirmed that DET induces ROS production in HCT116 cells.
To investigate the association between DET-induced ROS generation and autophagy, ultrastructural changes of the cells,p62 and LC3A/B-Ⅱ protein expression levels in the presence of a ROS scavenger, NAC, were investigated. In the presence of NAC,TEM analysis and AO staining revealed the absence of autophagic vacuoles and red acidic vesicles in DET-treated cells (Supplementary Figure 2). Correspondingly, NAC effectively suppressed the DETinduced accumulation of LC3A/B-Ⅱ, concomitant with an increase in p62 protein expression (Figure 4E and F). These findings indicated that DET-mediated autophagy is caused by the production of ROS in HCT116 cells. Furthermore, the activation of apoptosisrelated proteins by DET including cleaved caspase-3, cleaved PARP and DR5 proteins was suppressed while the procaspase-9 and -10 were increased in the presence of NAC (Figure 4G and H).
3.6. ROS-dependent MAPK pathway activation induced by DET treatment
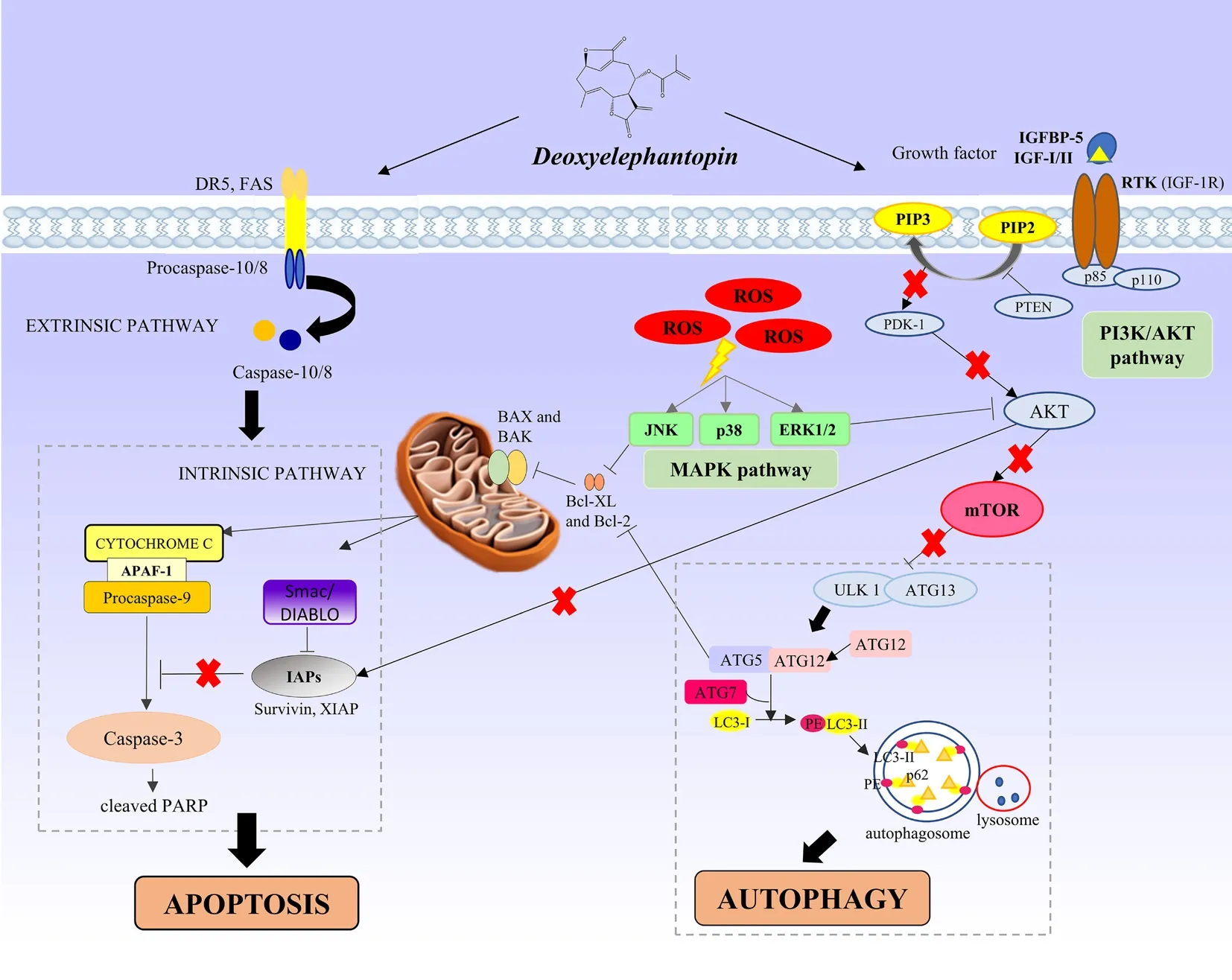
Figure 8. Schematic illustration of apoptosis and autophagy conferred by deoxyelephantopin (DET) via the activation of ROS-mediated MAPK and PI3K/AKT/mTOR signaling in colorectal cancer cells.
In the present study, it was observed that DET induced a timedependent increase in the expression of phosphorylated ERK1/2,JNK and p38 proteins (Figure 5A and B), indicating that the activation of MAPK is required for DET-induced apoptosis. We further investigated the potential of ROS in activating apoptosis via MAPK pathway triggered by DET. In the presence of NAC, the phosphorylation of ERK1/2, JNK and p38 MAPK proteins were inhibited (Figure 5C and D), which suggests that the activation of MAPK is ROS-dependent in HCT116 cells. To assess the involvement of MAPK pathway in DET-induced apoptosis, p38 specific inhibitor (SB202190), JNK specific inhibitor (SP600125)and ERK specific inhibitor (UO126) were used in combination with DET treatment. DET treatment significantly reduced the activation of caspase-3 and PARP in the presence of SB202190, SP600125 and UO126 inhibitors when compared to DET treatment alone (Figure 5E and F). Hence, when the MAPKs, i.e. p38, JNK and ERK were inhibited by their respective inhibitors, apoptosis induction by DET via the MAPK pathway was hampered. These findings suggested that DET-induced ROS-dependent apoptosis is mediated via MAPK pathway in HCT116 cells.
3.7. Suppression of PI3K/AKT/mTOR signaling pathway by DET treatment
The involvement of PI3K/AKT/mTOR on DET-induced autophagy and apoptosis in HCT116 cells was investigated. DET inhibited the phosphorylation of AKT at Ser473 and Thr308 sites timedependently (Figure 6). In addition, DET markedly reduced the expression of phosphorylated phosphatase and tensin homolog(PTEN) (inactivated form of PTEN) and pyruvate dehydrogenase kinase 1 (PDK1), indicating the activation of PTEN and inactivation of PDK1 by DET. The AKT downstream molecule, p-mTOR was also downregulated. In addition, treatment with DET significantly suppressed the expression of regulatory-associated protein of mTOR(RAPTOR) at 12 and 24 h (Figure 6). These data suggested that DET inhibits PI3K/AKT/mTOR pathway in HCT116 cells.
3.8. Inhibition of the growth of HCT116 cells in vivo by DET
The in vivo effect of DET on CRC was examined in mice tumor xenograft. As illustrated in Figure 7A, the body weights of all mice groups did not significantly change except for mice treated with 50 mg/kg of 5-fluorouracil, in which there was a significant reduction in tumor sizes and all mice died by day 10 likely from 5-fluorouracil toxicity associated with the dose used (Figure 7B). Tumor sizes were significantly reduced when the mice were treated with 1.25 and 2.5 mg/kg DET (Supplementary Figure 3). In the 2.5 mg/kg DET-treated group, the tumor volumes were significantly reduced by 73.48% after 28 days of DET administration (Figure 7B). In order to verify the involvement of apoptosis and autophagy as the underlying mechanisms of DET in the in vivo model, IHC staining was performed. It was notable that DET significantly upregulated cleaved caspase-3, LC3A/B and BAX protein expressions. In contrast, IHC analysis clearly demonstrated significant reduction of proliferating cell nuclear antigen (PCNA)-positive cells and Bcl-2 protein expression compared to the control group (Figure 7C and D).Hence, both in vivo and in vitro findings showed that DET induces colorectal cancer cell death via apoptosis and autophagy.
4. Discussion
DET has garnered a lot of attention as an effective anti-cancer agent owing to its anti-migration, anti-angiogenesis and anti-tumor effects[18,23,17]by targeting multiple signaling pathways important for cancer cell proliferation[24]. We previously reported that DET induces cell cycle arrest and apoptosis in HCT116 colorectal cells[20].Based on previous evidence of its effectiveness in suppressing growth of many cancer cell types, the present study aims to map the stepwise events leading to cell death by examining the interplay between apoptosis and autophagy using HCT116 as a model as well as to provide in vivo evidence for the effectiveness of DET in targeting CRC.
Apoptosis is often a major target in the development of novel cancer chemotherapy[25,4,26]. Results from the present study showed that DET induces apoptosis in HCT116 cells by DR5 and FAS receptors activation followed by caspase-10 and -8 and then caspase-3 activation. Moreover, caspase-9 and caspase-3 of the intrinsic pathway were also activated which led to apoptosis in HCT116 cells. These findings suggest that DET induces apoptosis via both the extrinsic and intrinsic apoptosis pathways in HCT116 cells. In line with the induction of apoptosis, dissipation of mitochondrial membrane potential was observed and evidenced by the upregulation of BAX and BAK, and the concomitant downregulation of Bcl-2. The Bcl-2 family members are known to govern mitochondrial pathway and initiate mitochondrial outer membrane permeabilization following excessive signals from the pro-apoptotic BAX/BAK over anti-apoptotic Bcl-2[27]. This shows that the oligomerization of BAX and BAK has inhibited Bcl-2 and resulted in the dissipation of mitochondrial membrane potential and eventually caused apoptosis in the DET-treated cells. IAPs family members such as XIAP and Survivin are direct inhibitors of caspases and consequently suppress apoptosis[28]. The release of the apoptogenic molecules including Second mitochondria-derived activator of caspase (Smac) and High temperature requirement protein A2 (HTRA2) enhances apoptosis via abrogation of IAPs generation and constitutive activation of caspase cascades. DET also attenuated the protein expressions of Survivin and XIAP, which have further propagated the activation of caspase-9 and caspase-3. These findings further provided evidence to support our previous preliminary study on DET inhibition of HCT116 cell growth through apoptosis and cell cycle arrest[20].
The intricate interplay between autophagy and apoptosis is reported[29], but how DET modulates the series of events leading to autophagy and apoptosis activation has yet to be reported.
Accumulation of autophagy proteins (LC3A/B-Ⅱ, ATG7 and ATG5)following treatment with DET was observed and this finding is in agreement with previous reports[30,31]. It was further determined that autophagy preceded apoptosis in HCT116 cells following treatment with DET. Treatment with z-VAD-FMK, a caspase inhibitor resulted in the accumulation of LC3A/B-Ⅱ, suggesting that upon inhibition of apoptosis, the DET-treated cells were instead maneuvered towards autophagy induction. This was further verified by the blockade of autophagy by autophagy inhibitor 3-MA, which resulted in reduced cleavage of caspase-3, indicating that the blockade of autophagy has inhibited apoptosis. In addition, using siRNA to knockdown LC3 to inhibit autophagy, we further confirmed in the absence of autophagy,apoptosis was inhibited as evidenced by increased cell viability and resistance of DET-induced apoptosis. Apart from DET, autophagydependent apoptosis induced by various other anti-cancer agents has been reported[32,33].
Anti-cancer agents such as oxaliplatin, capsaicin and curcumin are known to induce oxidative stress and apoptotic responses via ROS production[34-36]. In the present study, dose-dependent increase of ROS generation in HCT116 cells upon DET treatment was observed. In addition, there was an accumulation of AO-stained acidic vesicular organelles and autophagosomes, which occurred hours after the elevation of ROS in HCT116 cells. These findings confirmed that DET triggers excessive production of ROS in HCT116 cells, which appears to be an early event in the induction of cell death.
MAPK is a central signaling pathway that is involved in cell proliferation and differentiation, whereby, activation of JNK and p38 MAPKs generally promotes apoptosis while activation of ERK1/2 promotes cell proliferation[3]. However, there was new evidence that showed anti-cancer agents could prolong the activation of ERK1/2 and promote apoptosis in cancer cells. For instance, taxol has been shown to induce apoptosis via the activation of ERK in MCF7 cells[37]while hispolon from the fungus Phellinus linteus has been shown to induce apoptosis via the phosphorylation of ERK1/2, p38 MAPK and JNK1/2 pathways in human nasopharyngeal cancer cells[38]. Consistent with these findings, treatment of DET induced prominent activation of p38 MAPK, ERK and JNK which was evident at 12 h and peaked at 24 h in HCT116 cells. Furthermore,treatment of HCT116 cells with specific inhibitors for ERK(UO126), JNK (SP600125) and p38 (SB202190) as well as NAC prior to the exposure to DET attenuated DET-induced apoptosis as evidenced by the downregulation of cleaved PARP when compared to DET treatment alone. These results revealed that activation of MAPK signaling pathway is associated with DET-induced apoptosis in HCT116 cells.
PI3K/AKT signaling is implicated in malignant transformation,aberrant cell growth as well as drug resistance[8]. A growing number of studies have demonstrated that several naturally derived compounds target PI3K/AKT/mTOR signaling pathway.For instance, neoalbaconol induces multiple cell death pathways in nasopharyngeal cancer cells by inhibiting PI3K/AKT/mTOR signaling pathway[39]. Our findings revealed that DET-induced apoptosis could also be attributed to the inhibition of AKT phosphorylation as evidenced by the suppression of PDK1 and AKT phosphorylation at the site of Thr308/Ser473. Further downstream of the PI3K/AKT pathway is another mediator of cell survival, mTOR which consists of mTORC1 and mTORC2 with their respective regulatory proteins, RAPTOR and rapamycin-insensitive companion of mammalian target of rapamycin (RICTOR)[40]. Our current findings revealed that DET abrogated the phosphorylation of mTOR.mTOR can be activated by Ras homolog enriched in brain (Rheb).Rheb activation is directly inhibited by tuberous sclerosis 2 (TSC2)-TSC1 complex. Activation of ERK activates the TSC2-TSC1 complex whereas activation of AKT increases TSC2 phosphorylation resulting in the dissociation of the TSC2-TSC1 complex[41,42].Hence, increased ERK activation and decreased phosphorylation of AKT which led to the overall inhibition of mTOR following DET treatment could be mediated through the activation of TSC2-TSC1 and Rheb downregulation. In addition, inactivation of mTORC1 could also stimulate the formation of the Unc-51-like kinase 1 (ULK 1)-containing pro-autophagic complex which ultimately leads to the formation of autophagosomes[43]. Thus, findings from the present study support the involvement of PI3K/AKT/mTOR pathway in the induction of both apoptosis and autophagy by DET.
Thus far, the in vivo anti-tumor efficacy of DET on human colorectal cancer cells has yet to be reported. Through our mouse xenograft study, it is observed that DET significantly inhibited tumor growth and also enhanced the survival of mice. Importantly, DET did not cause significant weight loss in mice and produced no apparent sign of toxicity. These findings are in agreement to those of a previous toxicity study which demonstrated that the maximum tolerated dose for DET is 40 mg/kg in the mice[44]. Colorectal tumorigenesis is a result from dysregulation of apoptosis and aberrant cell proliferation which is often associated with proliferative proteins such as PCNA[45]. Notably, our in vivo data of DET inhibition of tumor cell growth is substantiated by the drastically lower PCNA labelling index compared to the control mice group as demonstrated by immunohistochemical analysis. The lower PCNA expression is also consistent with in vitro findings from our previous study whereby,DET inhibits cell proliferation through cell cycle arrest at S phase in HCT116 cells[20]. In addition, immunohistochemical findings also showed that DET reduced Bcl-2 and elevated BAX, cleaved caspase-3 and LC3 expressions which corroborated findings from our in vitro studies.
In summary, our study has demonstrated that DET from Elephantopus scaber L. exerts significant in vitro and in vivo anti-tumor effects in HCT116 human CRC through the generation of ROS and induction of autophagy-dependent apoptosis. DET induces ROS-dependent apoptosis and autophagy by targeting multiple pathways including MAPK and PI3K/AKT/mTOR (Figure 8). It is proposed that DETinduced autophagy precedes apoptosis, which is mediated in concert with the inhibition of PI3K/Akt/mTOR signaling and activation of ROS-dependent MAPK pathway (Figure 8). Taken together,the ability of DET to alter these prominent signaling pathways in controlling cancer cell growth signifies the enormous potential of DET to be developed as a multi-target therapeutic agent for colorectal cancer.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would also like to express their gratitude to Prof.Dr. Noor Hasima Nagoor Pitchai and Phuah Neoh Hun for their generosity and assistance in providing the indispensable laboratory facilities to complete the research.
Authors' contributions
CKC and KLT performed experiments, data analysis and interpretation. CKC drafted the manuscript. WPF and KLT performed critical revisions of the manuscript and prepared the final approved version for publication. HAK conceived the study and reviewed manuscript draft. All authors participated in experimental design.
 Asian Pacific Journal of Tropical Biomedicine2020年3期
Asian Pacific Journal of Tropical Biomedicine2020年3期
- Asian Pacific Journal of Tropical Biomedicine的其它文章
- Thai Perilla frutescens fruit oil alleviates carbon tetrachloride-induced hepatotoxicities in rats
- Syringic acid improves oxidative stress and mitochondrial biogenesis in the liver of streptozotocin-induced diabetic rats
- Effect of protease inhibitor from Agaricus bisporus on glucose uptake and oxidative stress in 3T3-L1 adipocytes