
Toll样受体家族介导疼痛和痒觉信号的分子机制*
2020-02-10 08:20白占涛王志红高永静
中国疼痛医学杂志 2020年1期
张 丽 白占涛 王志红 高永静 刘 通
(1江苏大学附属昆山市第一人民医院麻醉科,昆山215300;2延安大学生命科学学院,延安716000;3苏州大学神经科学研究所,苏州215123;4南通大学疼痛医学研究院,特种医学研究院,南通226019)
众所周知,疼痛和瘙痒是机体两种截然不同的主观感觉,但两者又彼此密切联系。国际疼痛研究会将疼痛定义为“一种与实际或潜在的组织损伤相关的不愉快的主观感觉和情感体验”[1]。目前,瘙痒的一般定义为“一种诱发抓挠欲望或反射的不愉快的躯体感觉”[2]。疼痛和瘙痒所诱发的均为躯体不愉快的主观感觉体验,但是疼痛引起的是退缩反射,而瘙痒引起的是抓挠反应[3]。疼痛可以发生在机体除大脑以外的深部和浅表几乎所有器官组织,而瘙痒仅发生在皮肤和粘膜等浅表组织处。在急性条件下,疼痛和瘙痒的感觉都可以作为警告信号,保护机体免受可能的伤害性刺激造成的损伤。急性瘙痒可以通过抓挠[4]和疼痛刺激得到缓解;而吗啡等止痛剂则常常引起机体的瘙痒感觉[5]。
在慢性疾病的条件下,不论是疼痛还是瘙痒的症状,都成为一种对机体无益且难以适应性的疾病状态,严重影响病人的睡眠和生活质量。有趣的是,在慢性疼痛的病理条件下,瘙痒刺激可以诱发疼痛感觉;而在慢性瘙痒的病理条件下,疼痛刺激也可以引起瘙痒感觉[6]。特别是在慢性病理条件下,瘙痒和疼痛的病理机制有着许多相似之处[6]。慢性疼痛和慢性瘙痒的临床症状也有许多相似之处。对慢性疼痛来言,包括外周或中枢神经系统损伤引起的神经病理性疼痛、外周组织损伤或炎症诱导的炎症性疼痛、功能障碍性疼痛、以及肿瘤引起的癌症痛等;其临床表现为自发性疼痛、痛觉过敏(指对阈上疼痛刺激的反应性增强)、触诱发痛(无害的轻触刺激诱发疼痛)[1]。慢性瘙痒可以在多种疾病状态下发生,包括如银屑病等皮肤疾病、胆汁淤积等肝脏疾病、慢性肾病、糖尿病等代谢性疾病以及带状疱疹病毒感染等,其临床表现为自发性瘙痒、痒觉过敏、触诱发痒(无害的轻触刺激诱发瘙痒)等[2]。
一、疼痛和瘙痒的神经传导通路
外周组织中瘙痒和疼痛的信号均可由初级感觉神经元游离的外周神经末梢(主要是Aδ和C类纤维)所探测。初级感觉神经元属于假单极神经元,其胞体位于背根神经节(DRG)和三叉神经节(TG)[2]。初级感觉神经元发出的中枢轴突末梢投射到脊髓(或延髓)背角区域,在这里进行瘙痒和疼痛的信号的第一级中枢的加工处理。初级感觉神经元中负责感受疼痛刺激的细胞亚群(即伤害性感受器;Nociceptor)中有一小部分专门负责感受瘙痒刺激,也称为痒感受器(Pruriceptor)。初级感觉神经元上表达多种类型的感受疼痛或瘙痒刺激的受体,包括G蛋白偶联受体(GPCR)、瞬时感受器电位离子通道(TRP通道)、酸敏感通道(ASICs)、细胞因子受体(如白细胞介素1β受体等)以及机械敏感离子通道(如PIEZO通道等)。其中,有些受体只感受疼痛刺激,如某些GPCRs、细胞因子或趋化因子受体等。有些受体既能感受疼痛也感受瘙痒,如组胺受体、蛋白酶激活受体(PARs)、五羟色胺(5-HT)受体某些亚型和缓激肽受体等。还有一些受体仅能感受瘙痒刺激,包括G蛋白偶联受体MrgprA3、5-HT7受体和胸腺基质淋巴细胞生成素(TSLP)受体等[2]。此外,初级感觉神经元进行疼痛和瘙痒的信号转导,也通常采用相互重叠的下游信号转导通路,如瞬时受体电位受体家族TRPV1、TRPV4和TRPA1通道等[7]。
位于脊髓的第二级投射神经元通过上行传导通路投射到脑干和丘脑,通过第三级神经元进一步投射到大脑皮层等相关区域,最终形成包含情绪、认知等多维度的痛觉感知。从脑干到脊髓的下行投射的5-HT能和去甲肾上腺素能神经纤维,通过激活脊髓局部环路的抑制性中间神经元[如γ-氨基丁酸(GABA)和甘氨酸],从而抑制疼痛信号的传导。关于痒觉的上行传导通路研究不多,一般认为跟痛觉的传导通路大致重合。近来的研究发现,臂旁核(parabrachial nucleus, PBN)是作为痒觉上行传导通路上的重要的中继核团[8]。此外,在中脑导水管周围灰质(periaqueductal gray, PAG),GABA能神经元的激活或谷氨酸能神经元的抑制均对瘙痒产生下行抑制的作用,而GABA能神经元还能编码痒觉的厌恶情绪[9,10]。最近的研究表明,中脑腹侧被盖区(ventral tegmental area, VTA)中的多巴胺能神经元的激活介导抓挠诱导的愉快感,而GABA能神经元的激活则介导痒觉诱导的厌恶情绪[11]。
慢性疼痛和慢性瘙痒的发病机制有许多相似之处,都包括外周初级感觉神经元的敏感性增强(称为外周敏化)和中枢神经系统的适应不良和敏感性增强(称为中枢敏化)[12]。例如,触诱发痛和触诱发痒是慢性疼痛和慢性瘙痒下的两种常见现象,而中枢敏化机制在其发病中均发挥重要作用[6]。在临床上,加巴喷丁和普瑞巴林等药物对慢性疼痛和慢性瘙痒均有较好的缓解作用[3]。最近的研究还发现,前脑GABA能神经元的脊髓移植对神经病理性疼痛[13]和神经病理性瘙痒产生了抑制作用[14],这一结果为脊髓抑制性突触传递丧失(即去抑制作用)在慢性疼痛和慢性瘙痒中的关键作用提供了重要的实验证据。
二、神经免疫互作信号参与疼痛和瘙痒信号的发生和加工
外周神经系统(PNS)和中枢神经系统(CNS)稳态的维持需要神经元和非神经元的免疫细胞之间的双向交流[15]。然而,这种神经免疫的相互作用在病理条件下会发生功能障碍,参与许多神经系统疾病的病理发生,包括中风、多发性硬化症(MS)、阿尔茨海默病(AD)、帕金森病(PD)和慢性疼痛[15]。机体在发生感染或组织损伤后,位于外周和中枢神经系统的多种非神经细胞类型,包括免疫细胞、角质形成细胞、神经胶质细胞、上皮细胞、癌细胞和入侵的细菌等,释放神经活性物质直接激活、敏化或沉默伤害性感受器或痒感受器,从而促进或抑制疼痛或瘙痒信号的产生[16]。虽然疼痛或瘙痒信号产生的分子机制可能不同,这个通用模型可作为一个有用的范式来描述各种非神经细胞类型以及疼痛或瘙痒介质的生物学功能。
Toll样受体(TLR)位于神经免疫系统的界面,并且神经元和非神经元细胞的TLRs激活后,通过直接地激活初级感觉神经元或促使非神经元细胞产生细胞因子或趋化因子,进而调节疼痛和瘙痒的信号加工过程[17]。本文重点讨论TLRs在调节疼痛和瘙痒信号中的新作用,并讨论了靶向TLRs信号转导通路可能作为缓解慢性疼痛和慢性瘙痒的新策略。
三、Toll样受体家族及信号转导通路
第一个被确定的TLR家族成员是果蝇的Toll分子。Toll对果蝇胚胎发育过程中的背腹模式以及抗真菌先天免疫反应至关重要[18]。哺乳动物的Toll样受体(TLR)随后被克隆,并被证明属于模式识别受体(PRRs),其通过直接识别病原体相关分子模式(PAMPs)和危险相关的分子模式(DAMPs)来启动先天免疫应答反应。免疫细胞中的TLRs参与会启动胞内信号传导途径,导致细胞因子和趋化因子的合成和释放[18]。TLRs介导的先天免疫应答也被认为是产生适应性免疫应答的先决条件。
迄今,已在人类和啮齿动物中鉴定出13种功能性TLRs[18]。TLRs是进化上保守的I型跨膜蛋白,并且包含介导配体识别的、富含亮氨酸重复序列的胞外结构域、跨膜区和激活下游信号传导的TIR结构域。根据它们的亚细胞定位,TLRs可分为细胞膜TLR (TLR1、TLR2、TLR4、TLR5、TLR6 和 TLR10)和内体TLR (TLR3、TLR7、TLR8和TLR9)。然而,根据细胞类型不同,一些内体TLRs(例如TLR7)也可以定位于细胞膜上(见表1)。 大多数类型的TLR在它们之间形成同源二聚体,而某些TLR则形成异二聚体,例如TLR1/TLR2和TLR2/TLR6。
TLRs检测来自微生物的不同PAMPs,包括病毒、细菌、分枝杆菌、真菌和寄生虫等[18]。对于细胞膜表达的TLRs,TLR1、TLR2和TLR6能够检测脂蛋白,TLR4能够检测脂多糖(LPS),TLR5负责检测鞭毛蛋白等。内体TLRs负责感知核酸,例如TLR3感知双链(ds) RNA,TLR7和TLR8感知单链(ss) RNA,TLR9感知CpG DNA。在病理条件下,TLRs可通过识别细胞坏死和组织损伤后释放的内源性DAMPs,来诱导无菌炎症反应[19]。TLRs的激活通过募集嗜中性粒细胞和激活巨噬细胞以及干扰素(IFN)刺激的基因表达,来清除侵入体内的病原体。此外,TLRs的激活还能导致树突细胞(DC)的成熟,这对于适应性免疫应答至关重要。
转接分子MyD88是大多数TLRs的下游信号转导所必需,但TLR3信号转导则不需要MyD88[18](见图1)。TLR2和TLR4也通过转接分子TIRAP转导信号(见图1)。TLRs识别配体后,MyD88募集IL-1R相关激酶(IRAK)。IRAK活化导致与肿瘤坏死因子受体相关因子6 (TRAF6)的相互作用,并导致NFκB (IκB)-激酶复合物(IKK复合物)的磷酸化。IκB的磷酸化导致IκB的降解,从而允许NFκB易位至细胞核并启动随后的基因转录。同时,TRAF6激活促分裂原活化蛋白激酶(MAPK)信号通路,包括细胞外信号调节激酶(ERK)、p38和c-Jun N末端激酶(JNK),并启动基因转录。TLRs信号通路的激活产生许多促炎细胞因子或趋化因子,包括TNF-α、IL-1、IL-6、IL-12、IL-8 和 MIP2 等,以及活性氧或活性氮(见图1)。TLR3和TLR4利用转接分子TRIF,最终激活NFκB和干扰素调节因子3 (IRF3)[18]。TRIF招募TRAF6用于NFκB的活化,类似于MyD88依赖性信号途径。TRIF信号传导导致IRF3的磷酸化及其核转位。IRF3的激活又导致I型干扰素(IFN)的转录。
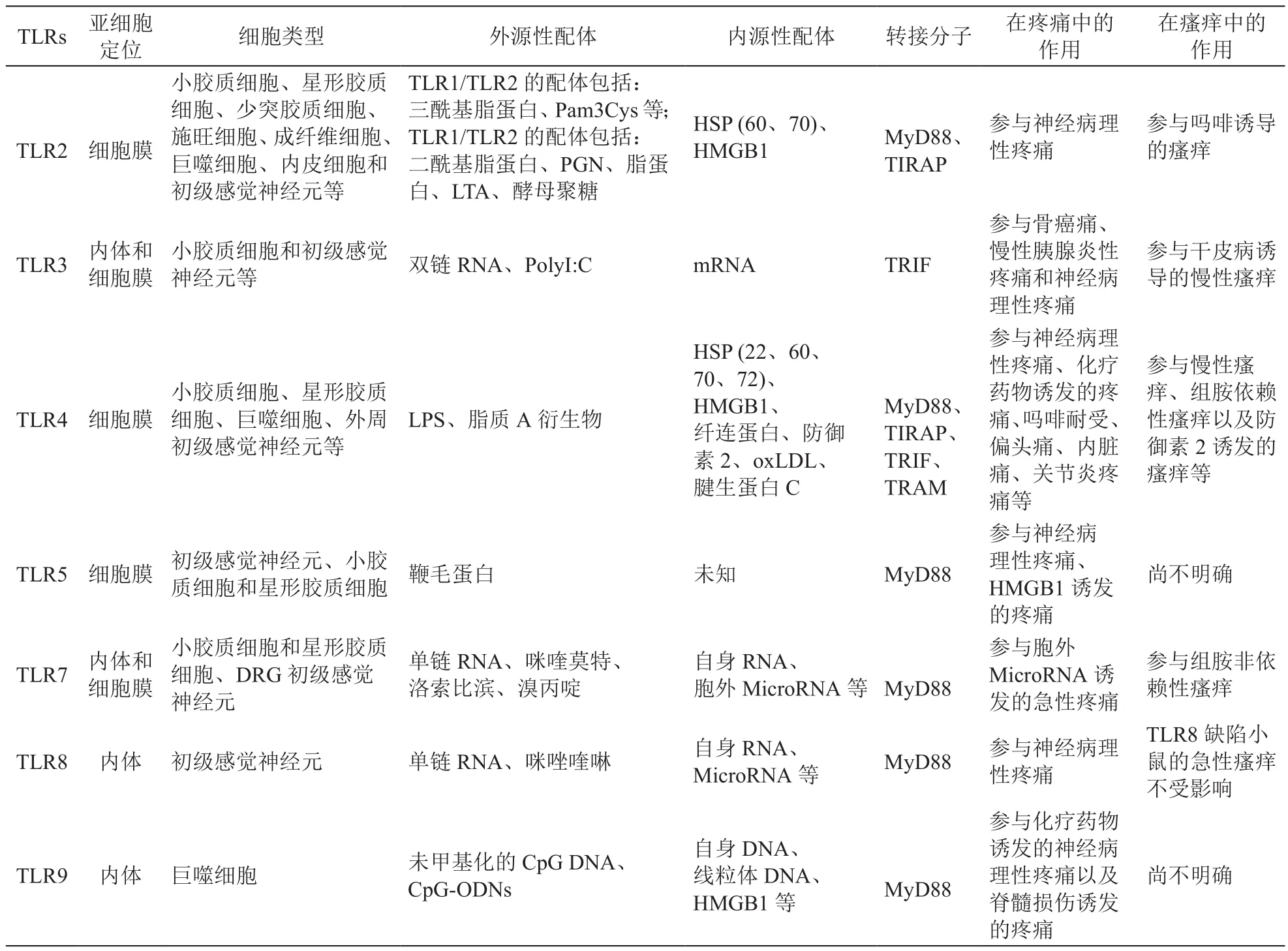
表1 Toll样受体家族概况、亚细胞定位、配体、衔接蛋白与疼痛或瘙痒
四、Toll样受体家族与疼痛
越来越多的证据表明,TLRs介导的神经炎症与慢性疼痛发病的外周敏化和中枢敏化机制有关。TLRs恰好位于神经免疫系统的界面,并且在外周神经系统(PNS)和中枢神经系统(CNS)中的多种细胞类型中表达[20]。为人们所熟知的是,非神经元细胞的TLRs的活化产生各种炎症介质,包括细胞因子、趋化因子和蛋白酶。近来最新的研究表明,在初级感觉神经元中,神经元TLRs的激活通过开放离子通道,并启动动作电位或使对致痛源和致痒源的神经元反应的敏感性增加。本部分综述了各种不同的TLR家族成员的功能及其在疼痛中的重要作用。
1.TLR2和疼痛
TLR2通过形成TLR2-TLR1和TLR2-TLR6异二聚体来区分识别多种脂肽。有证据支持一些DAMPs,例如HMGB1、HSP60、双糖链蛋白聚糖和透明质酸等,也可以被TLR2识别,尽管这些DAMPs并非仅由TLR2识别[21]。TLR2激活后通过转接分子MyD88依赖性途径启动细胞内信号传导。与脑中的其他细胞类型相比,TLR2的表达在小胶质细胞中高度富集,表明中枢神经系统中TLR2主要在小胶质细胞上表达[22]。还有证据表明,在星形胶质细胞、少突胶质细胞、施旺细胞、成纤维细胞、巨噬细胞、内皮细胞和初级感觉神经元中,也检测到TLR2的低水平表达。
TLR2主要通过在伤害感受信号通路中诱发神经炎症从而参与慢性疼痛的发生发展。与正常小鼠相比,外周神经损伤诱导的机械异常性疼痛和热痛觉过敏以及伴随的神经胶质细胞激活和脊髓炎症基因的表达,在TLR2基因缺陷小鼠中被抑制[23]。外周神经损伤后,在受损部位或DRGs出现巨噬细胞浸润,且TLR2的表达上调,可能参与外周神经炎症反应,例如TLR2介导的单核细胞趋化蛋白-1(MCP-1,也称为CCL2)表达的上调[24]。在临床研究中,与无痛病人相比,患有慢性疼痛的病人分离的外周血单核细胞对TLR2、TLR4和TLR7激动剂的反应有所增强[25]。近来,蛋白聚糖(aggrecan)的32个氨基酸残基的片段能直接激活背根神经节神经元产生MCP-1,参与骨性关节炎疼痛的发病过程[26]。这个结果提示,背根神经节神经元上表达的TLR2也参与外周神经系统的神经炎症过程(见图2)。
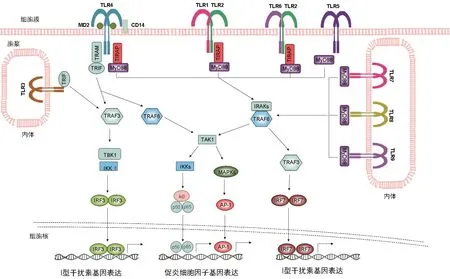
图1 哺乳动物免疫细胞中经典的TLRs信号转导示意图
2.TLR3和疼痛
TLR3识别双链RNA并通过TRIF途径启动信号,导致通过IRF3释放I型干扰素和通过NF-κB释放炎性细胞因子。关于TLR3与疼痛机制的研究相对较少。有研究表明,脊髓中TLR3的表达随着神经损伤后的小胶质细胞的活化或施加TLR3激动剂而表达升高[27]。通过鞘内注射反义寡核苷酸选择性敲低TLR3表达,能够减轻慢性胰腺炎后的机械性异常性疼痛[28]。TLR3基因缺陷小鼠也显示神经损伤诱导的异常性疼痛的缓解。然而,由TLR3所识别的内源性DAMPs,目前仍不清楚。
3.TLR4与疼痛
越来越多的证据表明,TLR4可能是促进与慢性疼痛发病机制相关的神经炎症反应的关键受体之一[29]。先前发现,TLR4广泛表达在小胶质细胞、星形胶质细胞和外周初级感觉神经元上。就神经元表达的TLR4而言,其主要在DRG和TG中的初级感觉神经元表达,可参与初级感觉神经元的激活或敏化,并直接促成疼痛信号传导的发生。TLR4在辣椒素敏感的小直径神经元上表达,与瞬时受体电位香草酸1 (TRPV1)和ATP门控嘌呤能受体P2X3共表达[30]。TLR4激动剂脂多糖(LPS)能够增强TRPV1+的感觉神经元的放电频率[30],因此通过TLR4依赖性机制直接影响初级感觉神经元的超兴奋性。有实验表明,初级感觉神经元的TRPA1也可能是LPS作用的另一个分子靶点[31]。由DRG的初级感觉神经元表达的神经元TLR4也通过使表达TRPV1的神经元敏感增强而参与紫杉醇诱导的外周神经病变及疼痛(见图2)[32]。
免疫细胞中TLR4的激活可以导致神经炎症,间接增强初级感觉神经元的兴奋性[29]。外周神经损伤或炎性后,脊髓中的TLR4的表达增加[33]。鞘内注射几种结构不同的TLR4拮抗剂LPS-RS(来自Rhodobacter sphaeroides的LPS)、(+)- 纳洛酮和(+)-纳曲酮(它们不结合于阿片受体),能够抑制神经损伤引起的机械疼痛超敏反应[21]。鞘内注射针对TLR4的siRNA或反义寡核苷酸以敲低TLR4的表达,可以减弱由外周神经损伤、骨癌和脊髓损伤诱导的痛觉超敏反应[21]。有趣的是,鞘内注射TLR4激动剂LPS在雄性小鼠中产生疼痛,而不是雌性小鼠中,表明TLR4参与神经性疼痛存在性别差异[34]。TLR4基因缺陷小鼠表现出由外周神经损伤、关节炎或自身免疫性神经病引起的痛觉超敏反应的显著降低[29]。有证据表明,神经胶质表达TLR4有助于吗啡诱导的神经炎症进而参与吗啡镇痛耐受的发生[35]。吗啡与MD-2的疏水核心结合(类似于LPS的脂质A的部分)并诱导TLR4寡聚化,导致NFκB介导的促炎性介质如IL-1β、TNF和一氧化氮的释放[36]。阿片类药物诱导的痛觉过敏(OIH)意味着阿片类药物暴露后导致机体对疼痛的敏感性增加。其中也涉及到小胶质细胞中的TLR4及其下游信号NLRP3炎性小体的参与[37]。化疗药物诱导的外周神经病变(CIPN)及疼痛是与化疗药物(如紫杉醇)的细胞毒性作用相关的严重不良后果,其特征是神经性疼痛可在化疗药物停止治疗后持续很长时间。研究表明,紫杉醇与MD2结合并诱导TLR4寡聚化,导致促炎反应[32]。因此,尽管需要更好地了解哪些条件对药理学靶向TLR4的反应最佳,但是TLR4受体拮抗剂可能有益于多种慢性疼痛疾病,包括神经性疼痛、炎性疼痛、OIH和CIPN。
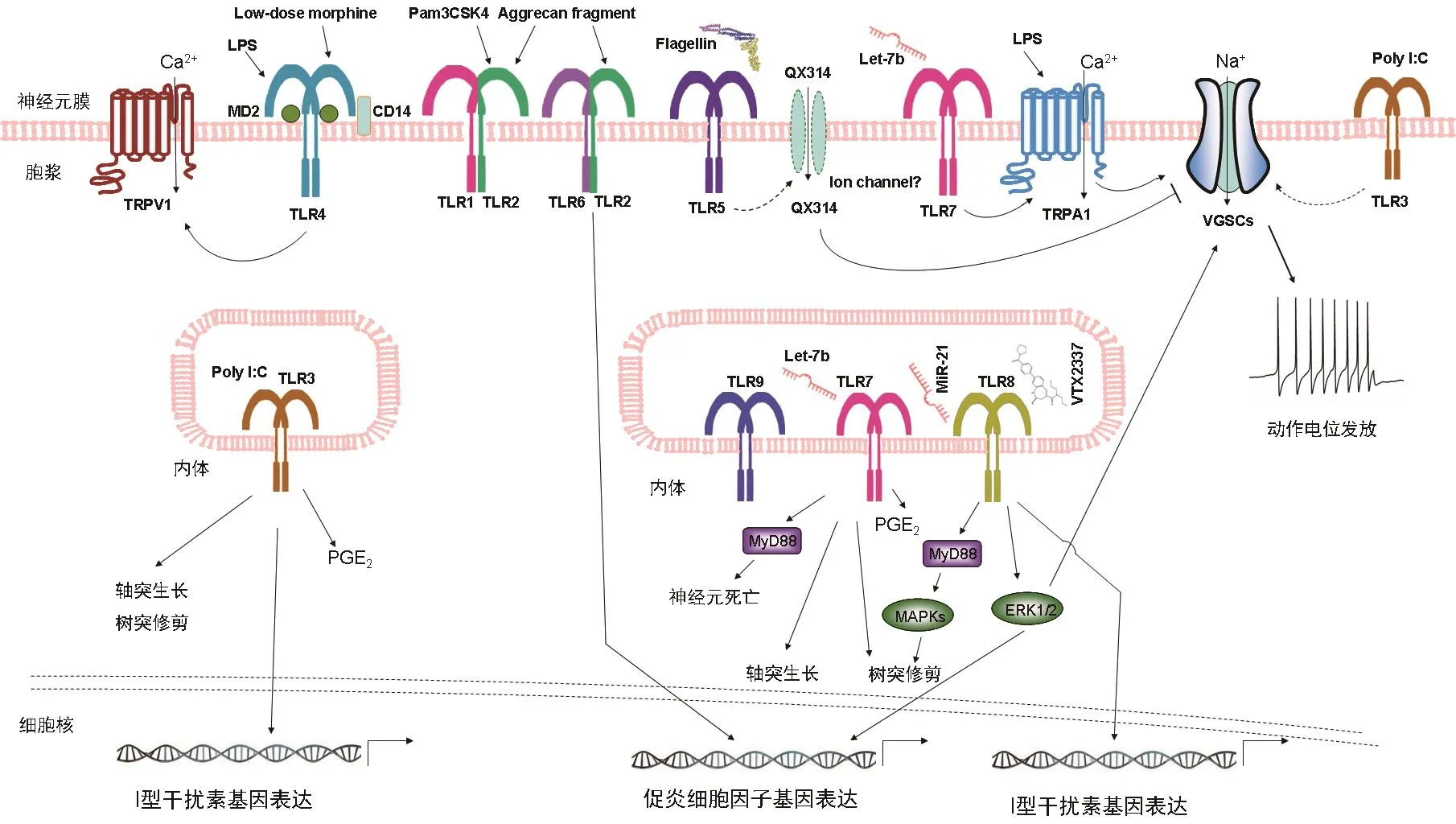
图2 哺乳动物神经元中非经典的TLRs信号转导示意图
TLR4识别的内源性DAMPs可能是TLR4介导的疼痛的罪魁祸首,尽管尚不清楚哪个DAMPs在此过程中是必需的。涉及哪些DAMPs可能取决于组织器官的不同。TLR4识别的几种可能的内源性DAMPs包括HSP60、HSP90、分泌到细胞外的HMGB1、生腱蛋白-C(属于细胞外基质糖蛋白)和半乳糖凝集素-3等[29]。有趣的是,最近的报道表明肠道微生物群也可能是TLRs配体的体内重要来源。早期研究发现,在缺乏肠道微生物群的无菌小鼠中,由炎性疼痛引起的机械疼痛超敏反应减少[38]。最近,在清除肠道微生物群后,化疗药物奥沙利铂诱导的机械痛觉过敏显著性降低,并且这些作用依赖于循环造血细胞上表达的TLR4[39]。近来发现,脑中内皮细胞上的TLR4识别肠道中革兰氏阴性细菌的LPS并导致脑海绵状血管畸形,而这种TLR4介导的病理可通过干预肠道微生物组来预防[40]。因此,推测靶向干预肠道微生物群,也能调节TLR信号传导的转导。
4.TLR5与疼痛
细胞膜TLR5识别细菌的鞭毛蛋白(flagellin),它是革兰氏阴性和革兰氏阳性细菌的结构蛋白。如图1所示,TLR5的二聚化导致转接分子MyD88依赖性的信号转导通路的激活。近来研究发现,TLR5在初级感觉神经元、小胶质细胞和星形胶质细胞中表达。原位杂交和免疫组织化学分析显示,TLR5主要表达在小鼠DRGs的中大型有髓鞘Aβ纤维上,它支配后爪无毛的皮肤并延伸到脊髓背角的深层[41]。TLR5也表达在人的DRG神经元中。L5脊神经结扎导致的机械痛敏在TLR5基因敲除小鼠中有所减弱。鞭毛蛋白激活神经元TLR5后,QX-314(一种膜不可渗透的利多卡因的衍生物)通过未知的一种通道进入神经元,导致钠电流的阻断,这主要发生在小鼠DRG的大直径Aβ纤维神经元中。鞭毛蛋白和QX-314的共同施加能够抑制化疗、神经损伤和糖尿病神经病变后的机械痛觉过敏。结果表明,TLR5介导的Aβ纤维沉默有望治疗神经性疼痛。近来的研究发现HMGB1可能作为TLR5的内源性配体候选者[42]。HMGB1在体外引发TLR5介导的MyD88信号级联,包括NFκB依赖性炎症介质的转录,例如TNF、IL-8和iNOS[42]。将鞭毛蛋白或HMGB1直接注射到大鼠的后爪中产生急性疼痛,其可被TLR5拮抗剂所抑制。因此,结果表明TLR5对细胞外内源性的HMGB1的识别,可能诱发痛觉过敏[42]。最近,过表达miR-150可以抑制TLR5介导的神经炎症反应,包括大鼠周围神经损伤后的COX-2、白细胞介素IL-6和TNF-α的表达,从而大大减轻了神经性疼痛[43]。总之,TLR5介导的神经免疫信号通过产生促炎介质而导致慢性疼痛。另一方面,初级感觉神经元中的神经元TLR5的激活也可能参与神经病理性疼痛的发生。
5.TLR7和疼痛
表达在内体上的TLRs通过感知核酸而促进先天免疫应答。这些内体中存在的TLRs被认为不同程度的表达在小胶质细胞、星形胶质细胞、少突胶质细胞、施旺细胞、成纤维细胞、内皮细胞和初级感觉神经元上。研究表明,培养的DRG神经元上表达TLR3、TLR7和TLR9,给予它们的特异性激动剂可增加TRPV1表达以及DRG感觉神经元的活性,并且能增加前列腺素E2的释放[44]。提示内体TLRs在DRG的初级感觉神经元中功能性表达。
TLR7识别单链RNA并启动MyD88依赖性途径,以释放NF-κB介导的炎性细胞因子或IRF7介导的I型IFN的转录(见图1)。尽管在小胶质细胞和星形胶质细胞中有低水平TLR7的表达,但是TLR7在DRG初级感觉神经元,特别是表达TRPV1的神经元上有表达[45]。最近的研究结果揭示了细胞外miRNA通过TLR7-TRPA1快速激发伤害感受神经元的新颖作用[46]。值得注意的是,miRNA let-7b与TLR7结合并激活TRPA诱导DRG神经元中快速内向电流和动作电位。这些电生理效应需要Let-7b的GUUGUGU基序[46]。足底内注射let-7b引起快速自发性疼痛,这些反应依赖于TLR7和TRPA1的表达[46]。因此,细胞外miRNAs可能作为内源性DAMPs,通过激活TLR7和TRPA1从而引发疼痛。
6.TLR8和疼痛
最近的研究发现,DRG上初级感觉神经元中的TLR8及其内源性配体miR-21共同导致小鼠的神经病理性疼痛的发生[47]。TLR8主要在中小型IB4+非肽能神经元中表达,并且在小鼠神经病理性疼痛的维持期间,TLR8阳性神经元的百分比有所增加。在Tlr8-/-小鼠中,神经损伤引起的机械痛敏和脊神经结扎(SNL)或紫杉醇(PTX)诱导的热痛敏显著降低。在Tlr8-/-小鼠中,SNL后脊髓中星形胶质细胞和小胶质细胞的活化减少与神经病理性疼痛行为的减少相一致。有趣的是,TLR8激动剂(VTX-2337)未激活DRG中的NF-κB。在鞘内注射VTX-2337后,p-ERK表达显著增加,而不是p38和JNK。在野生型(WT)和MyD88-/-小鼠中鞘内注射TLR8激动剂剂量依赖性地诱导机械痛敏,表明TLR8激动剂诱导的机械痛敏和ERK活化并不依赖于MyD88。
TLR8可识别单链RNA (ssRNA)和microRNA,并导致免疫细胞中促炎细胞因子的产生。Zhang等发现,miR-21的表达在神经病理性疼痛的条件下显著增加[47]。鞘内注射miR-21 可降低WT小鼠(而不是Tlr8-/-小鼠)的缩足阈值以及p-ERK 表达的增加,诱导DRG中TNF-α、IL-1β、IL-6、CCL2和CXCL1的表达增加。miR-21显著增加WT小鼠的DRG神经元中的动作电位的发放,但不增加Tlr8-/-小鼠的动作电位的发放。因此,miR-21在DRG中调节疼痛、激活ERK、产生炎症介质和增强神经元兴奋性的作用需要TLR8的介导(见图2)。
7.TLR9和疼痛
TLR9定位于内体中被DNA与未甲基化的CpG二核苷酸激活,并启动MyD88依赖性信号传导以激活转录因子NF-κB,从而产生促炎细胞因子和激活IRF7以产生IFNs。通过全身应用CpG ODN 2088阻断TLR9信号传导减少炎症信号传导并改善肿瘤诱导的热痛敏[44]和脊髓损伤诱导的热痛敏反应[48]。并且,TLR9拮抗剂或TLR9基因缺陷小鼠中,紫杉醇诱发的机械痛敏减弱[49]。紫杉醇导致巨噬细胞浸润到背根神经节(DRG),并且这种浸润不受TLR9基因缺陷的影响。紫杉醇处理巨噬细胞和DRG组织使TNF和CXCL1的表达上调。足底内注射紫杉醇激活的巨噬细胞引起机械痛敏。有趣的是,TLR9突变或用TLR9抑制剂仅在雄性动物中发挥作用。最终,TLR9的拮抗剂减少了雌性裸鼠(具有T细胞和B细胞缺陷的表型)中紫杉醇诱导的机械性痛觉过敏。总之,小鼠巨噬细胞中TLR9信号传导在化疗药物诱导的神经性疼痛的发病中,具有明显的性别差异[49]。但是,在慢性疼痛下,哪种内源性配体激活TLR9途径参与疼痛,仍不清楚。
8.Toll样受体的辅助蛋白和疼痛
新的证据表明,TLR的辅助蛋白和转接分子也参与慢性疼痛的发病过程。辅助蛋白CD14显著增强TLR4对LPS的识别,并促进TLR2-TLR6异二聚体的免疫应答。与野生型小鼠相比,CD14基因缺陷小鼠的神经损伤诱导的机械痛敏和热痛敏的反应显著降低。在CD14基因缺陷小鼠中,在中枢施用TLR4激动剂纤连蛋白、CNS创伤和缺血性中风后,小胶质细胞激活和单核细胞的募集有所减弱[21]。因此,CD14介导的信号传导可能参与TLRs对DAMP的识别以及组织损伤诱导的炎症反应。
转接分子MyD88是几乎所有TLR介导的信号传导所必需的(除TLR3外),也参与慢性疼痛的发病机制。外周神经损伤诱导伤害性途径中的MyD88表达,包括DRG和脊髓中磷酸化的NFκB,磷酸化的细胞外信号调节激酶(ERK)和IL-1水平升高[50]。鞘内注射MyD88同源二聚化抑制肽能够减弱机械痛敏和热痛敏。尽管在表达Nav1.8的初级感觉神经元中条件性敲除Myd88的小鼠中,基础痛阈和急性炎性疼痛未受影响,但在这些小鼠的慢性神经病和炎性疼痛显著减弱[50]。
虽然其他TLRs在慢性疼痛中的作用仍然未知,但我们推测,在多种病理条件下,不同的TLR的激活可能导致不同的持续性疼痛状态。总之,越来越多的证据表明,神经元和非神经细胞中的TLRs及其相关的信号网络参与慢性疼痛的发生发展。靶向TLR信号转导通路有可能成为缓解慢性疼痛的新策略。
五、Toll样受体和痒觉
新的研究表明,TLRs对于瘙痒的发生发展也是至关重要的。TLRs激活产生疼痛或瘙痒,可能取决于入侵的外源性PAMPs和内源性DAMPs的性质。
1.TLR3和痒觉
研究发现,TLR3可能是介导小鼠慢性瘙痒的突触传递和中枢敏化的关键调节因子[51]。TLR3表达在TRPV1+以及GRP+的DRG的小直径初级感觉神经元中,而且还被转运至投射到脊髓背角的中枢轴突末端。TLR3激动剂可以诱导DRG神经元产生内向电流和动作电位。TLR3激动剂在WT小鼠中引发抓挠,但不引起TLR3基因敲除小鼠的抓挠行为。此外,脊髓背角II层神经元的兴奋性突触传递和脊髓的长时程增强(LTP)现象在TLR3基因敲除小鼠中受损[51]。此外,在TLR3基因敲除小鼠中,组胺依赖性和非依赖性急性瘙痒和皮肤干燥诱发的慢性瘙痒显著降低。因此,TLR3可通过增加外周初级感觉神经元的兴奋性和增强脊髓中的脊髓突触传递来调节瘙痒的中枢敏化过程。但参与瘙痒的TLR3的内源性配体尚不清楚。
2.TLR4和痒觉
越来越多的证据表明,神经元和非神经元中TLR4对急性和慢性瘙痒也有重要作用。尽管TLR4表达在TRPV1+的DRG感觉神经元上,但DRG神经元中LPS对TLR4的激活不会引发内向电流和动作电位[52]。皮内注射TLR4激动剂LPS也不能诱导小鼠的急性瘙痒。在TLR4基因敲除小鼠中,组胺诱导的急性瘙痒减少,并且组胺诱导的感觉神经元中的细胞内钙信号和内向电流在TLR4基因缺陷的小鼠也显著减少[53]。在TLR4基因缺陷的DRG神经元中,组胺的敏感性降低可能是由于TRPV1活性降低所致[53]。因此,初级感觉神经元表达的TLR4通过增强TRPV1活性可增强组胺诱导的瘙痒信号转导,但它不足以诱导急性瘙痒。
近来的研究发现,由星形胶质细胞表达的TLR4被认为在慢性瘙痒中起关键作用,但不参与急性瘙痒[52]。干皮病、接触性皮炎和过敏性接触性皮炎相关的慢性瘙痒,在TLR4基因敲除小鼠显著减少[52]。鞘内注射TLR4拮抗剂LPS-RS不影响急性瘙痒,但抑制干皮病诱导的慢性瘙痒。干皮病能使TLR4的表达持续上调,并且能诱导脊髓背角中表达GFAP的星形胶质细胞中TLR4的表达增加。有趣的是,慢性瘙痒引起的抓挠在脊髓星形胶质细胞增生中起着重要作用[52]。另一项研究表明,由信号转导和转录激活因子3 (STAT3)信号传导可以介导慢性瘙痒诱导的反应性星形胶质细胞增殖[54,55]。Lipocalin-2 (LCN2)同时被鉴定为星形胶质细胞STAT3依赖性的免疫因子,对慢性瘙痒的发生发展至关重要[55]。这些发现表明,由TLR4和STAT3信号传导介导的脊髓星形胶质细胞的激活,对于慢性瘙痒的发展起关键作用。
最近的研究表明,抗菌肽人源β-防御素2 (hBD2)在银屑病病人中的表达大幅增加[56]。在小鼠中,hBD2诱导了瘙痒反应,并且与野生型小鼠相比,hBD2在TLR4敲除小鼠中诱导的瘙痒显著减少[56]。此外,皮肤的免疫细胞(而非DRG神经元表达的)TLR4介导小鼠中hBD2诱导的瘙痒[56]。因此,该结果表明,皮肤细胞表达的TLR4(可能是巨噬细胞),可能在慢性瘙痒中起关键作用。
3.TLR7和痒觉
TLR7被证明由DRG初级感觉神经元表达,其与TRPV1、胃泌素释放肽(gastrin releasing peptide, GRP)、TRPA1和 MrgprA3共表达[45]。皮内注射TLR7的配体咪喹莫特或洛索立宾,剂量依赖性诱导小鼠抓挠行为。在急性分离的小鼠DRG神经元中,TLR7配体咪喹莫特和洛索立宾引发快速内向电流和动作电位,表明TLR7配体可直接激活DRG神经元。与野生型小鼠相比,TLR7基因敲除小鼠表现出正常的热和机械疼痛,并且炎症和神经性疼痛的行为也未改变[45]。值得注意的是,TLR7基因敲除小鼠显示出对组胺非依赖性瘙痒的显著减少,包括氯喹、内皮素-1和SLIGRL-NH2等。因此,TLR7对于组胺依赖性瘙痒是必需的,但对于急性疼痛的发生可能并不必要。
值得注意的是,动物行为、膜片钳记录和钙成像的研究揭示了令人惊讶的结果,咪喹莫特诱导的DRG神经元的活化与TLR7无关[57]。最近的研究进一步发现,咪喹莫特是一种直接的但较弱的TRPA1激动剂,可激活一部分表达TRPA1的神经元,TRPA1是咪喹莫特诱导的DRG神经元激活所必需的,也是咪喹莫特诱导瘙痒行为的原因[58],这些数据表明,初级感觉神经元表达的TLR7可能并不是咪喹莫特诱导的小鼠瘙痒所必需的。最近发现,分泌的细胞外miRNA (let-7b)作为一种新的疼痛介质,可通过激活伤害感受神经元中的TLR7和TRPA1起作用[46]。鉴于疼痛刺激可以在慢性瘙痒条件下转变为瘙痒,miRNA let-7b也可能作为TLR7的新型内源性配体参与慢性瘙痒。总之,病理条件下释放的内源性配体可能通过激活神经元或非神经元TLR7受体,进而参与瘙痒信号的产生。
六、结论及临床意义
尽管大量的结果表明,神经元和非神经元中TLRs介导疼痛和瘙痒信号的产生,提示TLRs可能代表一种缓解慢性疼痛和慢性瘙痒的新型治疗靶点。但是,我们仍需要考虑以下几个关键问题。首先,大多数研究使用的是全身性TLRs基因敲除的动物,导致TLRs在所有细胞类型中缺乏,因此很难将行为学表型归因于特定细胞类型,例如免疫细胞、神经胶质或神经元。不同类型的细胞中TLRs在疼痛和瘙痒中的确切作用,需要应用条件性TLR基因敲除动物。其次,相同的TLR在不同类型的细胞上具有不同的信号转导通路。例如,神经元TLRs的细胞内信号转导与非神经元TLR的信号转导就明显不同。再次,鉴于TLRs广泛表达并且具有正常生理功能,包括启动先天免疫应答发应,故TLRs拮抗剂可能会产生不良反应。选择性靶向TLRs可能比广泛抑制更具有优势,以避免一些潜在的不良反应。最后,在组织或神经损伤或皮肤损伤后释放的TLRs的内源性配体是什么?这些内源性TLR配体在不同的病理性疼痛和瘙痒症状下,作用于神经元或非神经元细胞的具体机制是什么?TLRs识别内源性配体的分子结构基础的阐明,最终可通过改进的药物设计产生更好的药物治疗效果。
包括炎性疼痛和神经性疼痛在内的各种慢性疼痛,影响全世界大约15亿人。慢性瘙痒也是与皮肤病、全身性疾病和代谢紊乱相关的常见临床问题。慢性疼痛和瘙痒大大降低了人们的生活质量,而临床的治疗效果不尽如人意。本文中综述了Toll样受体介导疼痛和瘙痒信号的分子机制。阐明TLRs介导疼痛和瘙痒信号的分子机制,将为治疗疼痛和瘙痒提供潜在的治疗新靶点。
猜你喜欢
中国实用神经疾病杂志(2022年3期)2022-11-28
世界中医药(2022年17期)2022-10-15
昆明医科大学学报(2022年2期)2022-03-29
心血管病学进展(2022年2期)2022-03-21
中老年保健(2021年3期)2021-12-03
电子产品世界(2021年8期)2021-01-16
中国计算机报(2019年49期)2019-02-07
中国新闻周刊(2017年36期)2017-10-21
创新时代(2016年8期)2016-10-21
飞碟探索(2015年11期)2015-09-10
