
周期密度泛函理论研究1,3-二醚类内给电子体在β-MgCl2(110)面吸附过程
2020-02-09 02:03张平生黄安平谢克锋
广州化工 2020年1期
张平生,李 磊,高 琳,黄安平,谢克锋
(1 中国石油兰州化工研究中心,甘肃 兰州 730060;2 兰州市第六十五中学,甘肃 兰州 730070; 3 兰州交通大学化学与生物工程学院,甘肃 兰州 730070)
现今聚烯烃工业生产最广泛使用的Ziegler-Nata载体型高效催化剂是以MgCl2或Mg化合物制备的MgCl2载体高效催化剂或MgCl2/SiO2复合载体高效催化剂[1-3]。MgCl2能作为较为理想的载体是由其自身的结构特点所决定的,MgCl2具有相似于TiCl3的层状晶体结构,并且Mg2+的离子半径(0.65 Å)和Ti4+的离子半径(0.68 Å)很相近,Ti4+较易嵌入MgCl2晶格,在载体表面形成稳定的复合物MgCl2/TiCl4。在丙烯聚合过程中,为了提高聚丙烯的等规度,需要在催化剂制备过程中加入内给电子体[4-6]。内给电子体能够提高聚丙烯催化剂的机理是通过内给电子体占据MgCl2表面的无规活性中心位点,TiCl4占据等规活性位点,从而提高了聚丙烯的等规度。Ziegler-Natta催化剂被广泛应用60多年,但是其详细聚合机理还是不清楚,近些年,分子模拟方法应用于催化剂聚合机理的研究[7-15]。在国内北京化工研究院分子模拟研究室将分子模拟技术应用到茂金属催化剂开发中[16-18]。随着二维材料如石墨烯、MOS2和BN等的发展,周期性密度繁华论文成为研究二维周期性材料有力手段,它将材料有限团簇计算扩展到了无限,使材料模拟研究更加有效和科学。本文采用周期性密度泛函理论研究1,3-二醚类内给电子体在MgCl2(110)面无规活性中心的吸附过程,从分子尺寸解释其微观机理。
1 模拟策略和模拟方法
1.1 模拟策略
将给电子体1,3-二醚吸附于β-MgCl2(110)面上面,其中O原子与β-MgCl2(110)面Mg原子的进行配位(图1所示),利用量子力学方法将模型优化,得到能量最低的吸附状态。并且将纯粹的β-MgCl2和1,3-二醚分子模型优化,得到最低的能量。通过吸附前后能量差计算吸附能:
ΔE=EMgCl2/donor-EMgCl2-Edonor
其中EMgCl2/donor,EMgCl2,Edonor分别是吸附后体系的能量,纯的β-MgCl2(110)面和单分子给电子体的能量。

图1 β-MgCl2(110)面模型(a)及选取的1,3-二醚给电子体结构式(b)和立体结构(c)
1.2 模拟方法
本文的模拟计算是采用Material Studio 5.5软件中基于密度泛函理论的DMol3模块优化结构,在研究1,3-二醚与β-MgCl2(110)表面之间的化学相互作用时,由于需考虑二者之间电子相互作用的影响,因此,选用量子力学从头计算方法进行研究,在本计算几何优化过程中基组采用GGA-BLYP,k点为5×5×1,收敛精度为能量误差小于10-5Ha。
2 结果与讨论
2.1 结构优化
β-MgCl2为…AAA…层状结构,在其晶体中镁原子与周围6个氯原子配位,在(110)面,镁原子只与4个氯原子配位。镁原子处于缺电子状态,1,3-二醚上氧原子很容易与镁原子配位,占据MgCl2(110)面的活性位点。1,3-二醚在MgCl2(110)面结合方式有5种(图2所示):单点配位(a),单层双点配位(b),螯合配位(c),双层双点配位(d),错位双层双点配位(e)。我们研究5种结合方式的吸附能和相应的Mg-O距离。表1结果表明螯合配位具有最大的吸附能,因此具有最大的稳定性,这是由于螯合体系具有最稳定的结构,双层双点配位,错位双层双点配位也具有较大的吸附能,因而它们结构也是稳定的,但是单点配位和单层双点配位吸附能是正直,说明在这两个体系1,3-二醚与MgCl2(110)面存在排斥作用,这是由于在两类体系中甲氧基正对与MgCl2(110)面,甲氧基位阻很大,阻碍了氧原子与镁原子的相互吸引。同理,在螯合配位体系中位阻最小,因此,它有最小的Mg-O距离和最大的吸附能,为最稳定体系。为了详细理解1,3-二醚在吸附过程中的结构的变化,主要角度的变化我们进行了研究,图1(b)所示为研究的主要角度示意图。表2列出了5种构象的主要角度,计算结果表明,在形成所有稳定结构中,相比独立的1,3-二醚结构,在MgCl2(110)吸附的1,3-二醚分子中,∠C-O-C均增大,从111°增加到了113°,说明1,3-二醚分子有略微的伸直,这是由于端基甲氧基中氧原子与MgCl2(110)面氯原子形成弱的H…Cl氢键,减小了1,3-二醚分子至MgCl2(110)面距离,同时Mg…O相互作用增强。
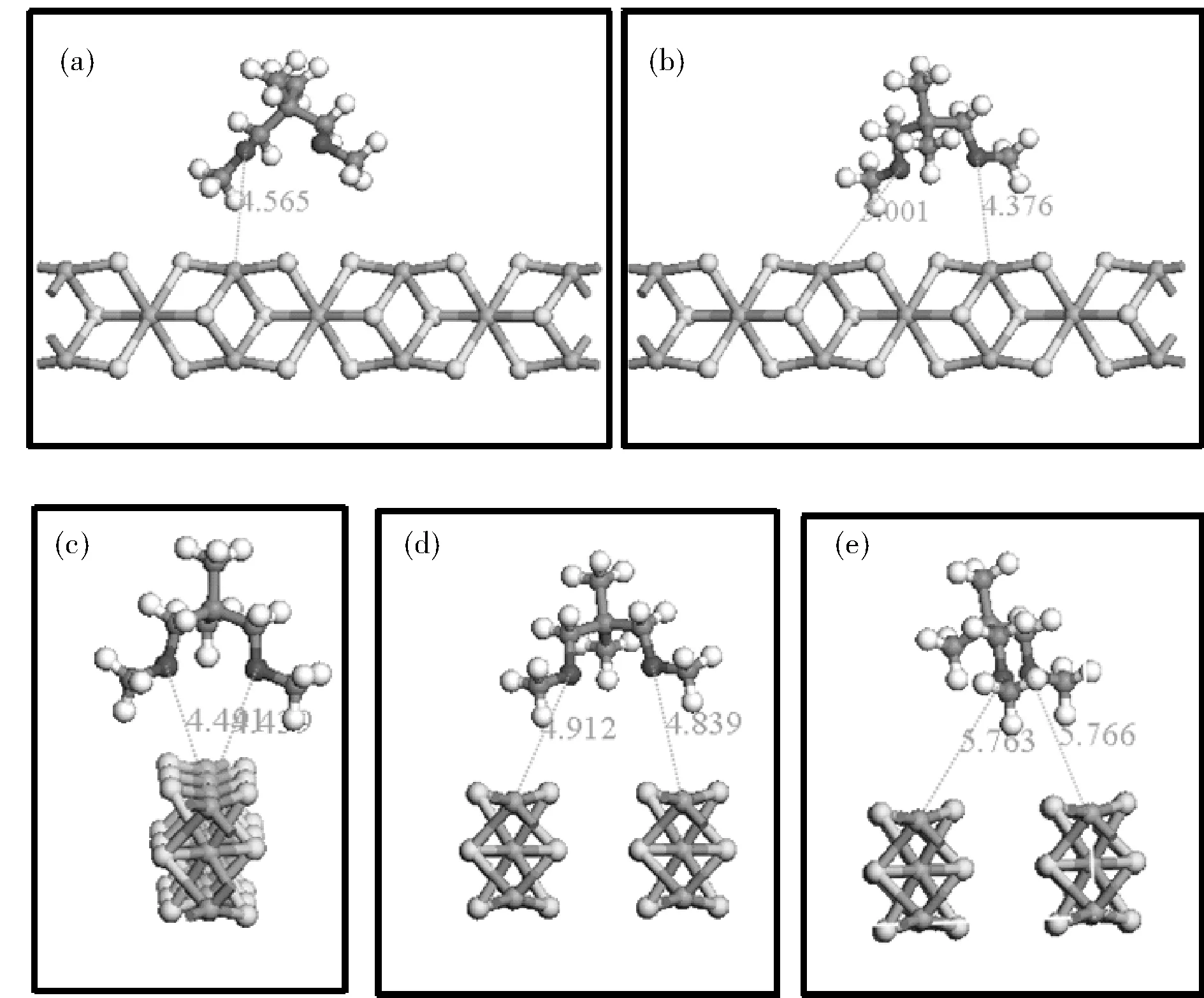
图2 1,3-二醚在MgCl2(110)面5种配位结构

表1 5种构象的吸附能和Mg-O距离

表2 纯粹和5中构象中1,3-二醚的主要角度(度)
2.2 旋转效应
螯合配位体系具有最稳定的结构,对于1,3-二醚螯合体系在MgCl2(110)面旋转构象进行了研究。在旋转过程中,我们选择了三个体系进行计算。在MgCl2(110)面,镁原子为四配位,镁原子周围有四个氯原子,以镁中中心组成一个平行四边形,两条对角线为Cl-Mg-Cl,我们三个计算体系为为1,3-二醚平面与MgCl2(110)面ab面垂直(a)和分别于两条对角线Cl-Mg-Cl平行(长边平行为图3(b),短边平行为图3(c))。表3为其吸附能和Mg-O距离计算结果,结果表明在b构象体系具有最到的吸附能,在此体系1,3-二醚平面与长对角线Cl-Mg-Cl平行,1,3-二醚中甲氧基上氢原子较好的能与MgCl2(110)面形成氢键,以至于体系能量降低,吸附能增大,1,3-二醚平面与短对角线Cl-Mg-Cl平行,Mg-O距离为4.77 Å和4.527 Å,由于O…Cl存在较大排斥,吸附能降低。
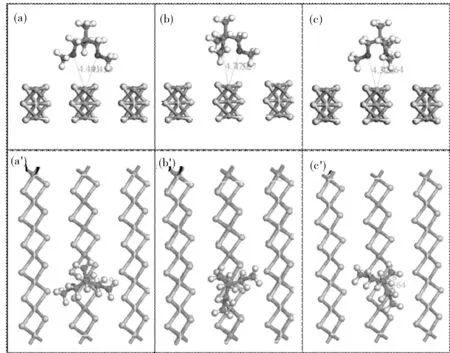
图3 1,3-二醚在MgCl2(110)面螯合配位旋转的三种构象

表3 螯合配位中3种构象的吸附能和Mg-O距离
3 结 论
本文采用周期性密度泛函理论研究了给电子体1,3-二醚分子在MgCl2(110)的详细吸附过程,研究结果表明1,3-二醚分子在MgCl2(110)可形成螯合配位,双层双点配位和错位双层双点配位三种稳定的构象,其中螯合配位吸附能最高,为最稳定构象,同时螯合配位方式1.3-二醚分子在MgCl2(110)面上的能够旋转,在1,3-二醚分子平面与长边Cl-Mg-Cl平行时,具有最大的吸附能,最稳定的构象。
猜你喜欢
同位素(2022年6期)2022-12-30
物理学报(2022年10期)2022-06-04
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
山东科学(2020年3期)2020-06-11
唐山师范学院学报(2020年6期)2020-04-16
中国食品学报(2019年10期)2019-11-12
唐山师范学院学报(2019年3期)2019-06-18
当代陕西(2019年6期)2019-04-17
农产品加工(2019年5期)2019-04-12
中国洗涤用品工业(2015年2期)2015-02-28
