
双子叶植物共同祖先基因组的拼接及应用
2020-01-19 06:09赵毓昊王金朋王希胤
唐山师范学院学报 2019年6期
赵毓昊,王金朋,王希胤
生命科学与技术
双子叶植物共同祖先基因组的拼接及应用
赵毓昊,王金朋,王希胤
(华北理工大学 生命科学学院,河北 唐山 063210)
以葡萄基因组数据为基础,结合双子叶植物7条祖先染色体的推断过程,重新整合拼接了双子叶植物共同祖先全基因组加倍前后的氨基酸和核苷酸序列。用拼接后的双子叶植物共同祖先基因组数据作为参照物种,选取毛果杨作为双子叶植物的代表进行基因组比较研究,结果显示,拼接后的双子叶植物共同祖先作为参照物种更加有利于双子叶植物中的比较基因组学研究。
祖先染色体;比较基因组学;葡萄基因组;参照物种
比较基因组学分为种间比较基因组学与种内比较基因组学,是在全基因组测序结果的基础上,通过比较已测序的基因组序列和基因组结构,获得物种基因组的差异信息,推断所比较基因的功能种类、表达过程以及和物种进化过程的关系。通过物种间或物种内的全基因组序列比对,还可以帮助预测基因位置、定位基因进而研究推断物种的进化过程。由现代基因组重构出祖先染色体核型进化过程,探索基因和染色体功能进化的机制,也都需要物种间的基因组信息比较才能实现[1,2]。
现有研究表明,双子叶植物共同祖先共有7条染色体[3],约1亿年前,双子叶植物祖先经历了一次全基因组三倍乘事件[4]。已被测序的拟南芥、葡萄、蔷薇科植物(苹果、草莓、桃、梅花)、杨柳科植物(毛果杨)基因组信息均揭示了这些双子叶植物起源于一个共同的六倍体祖先[5,6]。物种基因组经历多倍化之后发生了染色体数目加倍、复杂的染色体重排、部分同源染色体间的重组、大量基因丢失等遗传过程[7,8],因此,现代物种基因组结构十分复杂。
在已测序的双子叶植物中,葡萄基因组进化速度较慢,祖先遗传信息的保留相对完整,因此,在重构过程中常被作为参考基因组[9]。但研究中发现,多数双子叶植物与葡萄进行基因组比较时,染色体结构差异较大,在可视化的同源结构点阵图中的同源片段非常散乱,这给重构过程造成了困难。
JAILLON O等人根据葡萄基因组信息推断出了双子叶植物共同的祖先的基因组,但没有给出祖先基因组的遗传序列信息,所以,双子叶植物的比较基因组学研究中,仍选取葡萄为参考物种[3]。
本文以葡萄基因组数据为基础,结合双子叶植物7条祖先染色体的推断过程,整合、拼接了双子叶植物共同祖先加倍前后的氨基酸和核苷酸序列。分别以双子叶植物祖先加倍后的基因组和葡萄基因组作为参考基因组,绘制毛果杨基因组和参考基因组的同源结构点阵图,评价双子叶植物共同祖先基因组作为参考基因组的合理性。
1 实验部分
1.1 数据来源与处理
从公共数据库JGI(https://genome. jgi. doe. gov/portal/)下载研究所需的葡萄、毛果杨、毛果杨的核苷酸序列(CDS文件)、氨基酸序列(PEP文件)以及基因在染色体上的位置相关注释文件(GFF文件)。对下载的数据进行预处理,删除大量多余的基因组注释信息。
1.2 数据处理软件
利用BLAST软件做基因信息比对[10],搜索共线基因对。利用perl语言编写代码,绘制同源结构点阵图。
1.3 祖先基因组数据整合拼接过程

图1 葡萄19条染色体同源关系
参考相关文献[3],绘制葡萄19条染色体的同源关系,如图1所示。
图1中共有7种颜色,表示双子叶植物的7条祖先染色体,几乎每一种颜色都出现了3次,这说明双子叶植物的共同祖先发生了一次全基因组三倍乘事件。
1.3.1 加倍前的7条祖先染色体基因组数据整合过程
从葡萄的19条染色体中,每种颜色选一条完整且基因保留数量最多的染色体作为祖先染色体。本文选取葡萄的1、2、5、8、9、12、18号染色体作为双子叶植物的7条祖先染色体,并对这7条染色体的基因位置文件进行重新命名、排序。这7条染色体的氨基酸序列与核苷酸序列则与基因位置文件的新基因名保持一致。
1.3.2 加倍后的21条祖先染色体基因组数据拼接过程
比较完整的染色体如2、15、16号同源染色体以及6、8、13号同源染色体的处理方法与上步骤一致;而发生重组、交叉互换的染色体,如14号染色体,则需要借助同源片段搜索软件ColinearScan[11]搜索葡萄14号染色体与葡萄14号染色自身的同源片段,借助搜索到的同源片段信息找出染色体发生交叉互换的基因位置,体依照图示的颜色将其分割为两条染色体;同理切割葡萄的4号和7号染色体。
从图1中可见,深绿色出现了4次而不是3次,这说明代表深绿色的祖先染色体三倍乘后产生的3条子染色体中,其中一条发生了断裂。在葡萄染色体中,2号、18号都是完整的染色体,因此推断4号、7号染色体的深绿色部分是由一条染色体断裂而来的,应把深绿色部分连接在一起组成一条染色体,因此总共得到了21条染色体,这与预期一致。
2 结果与讨论
图2为葡萄与毛果杨物种之间的基因组同源结构点阵图。其中,横轴表示葡萄基因组,葡萄基因组的19条染色体按照从左向右的顺序进行排列。图中纵轴表示的是毛果杨的全基因组数据,毛果杨基因组的19条染色体按照从上而下的顺序进行排列。图中的每一个点表示BLAST软件搜索到的一对同源基因,匹配最佳的同源基因对用红色点标识出来,匹配较好的用蓝色点标识,其它同源基因对则用灰点标识。
图3为双子叶植物共同祖先与毛果杨物种之间的基因组同源结构点阵图。其中,横轴表示双子叶植物共同祖先基因组,ABCDEFG分别表示双子叶植物的7条祖先染色体,A1A2A3表示双子叶植物祖先全基因组三倍乘事件产生的三套子基因组,图中纵轴表示的是毛果杨的全基因组数据,毛果杨基因组的19条染色体按照从上而下的顺序进行排列。图中的每一个点表示BLAST软件搜索到的一对同源基因,匹配最佳的同源基因对用红色点标识出来,匹配较好的用蓝色点标识,其它同源基因对则用灰点标识。
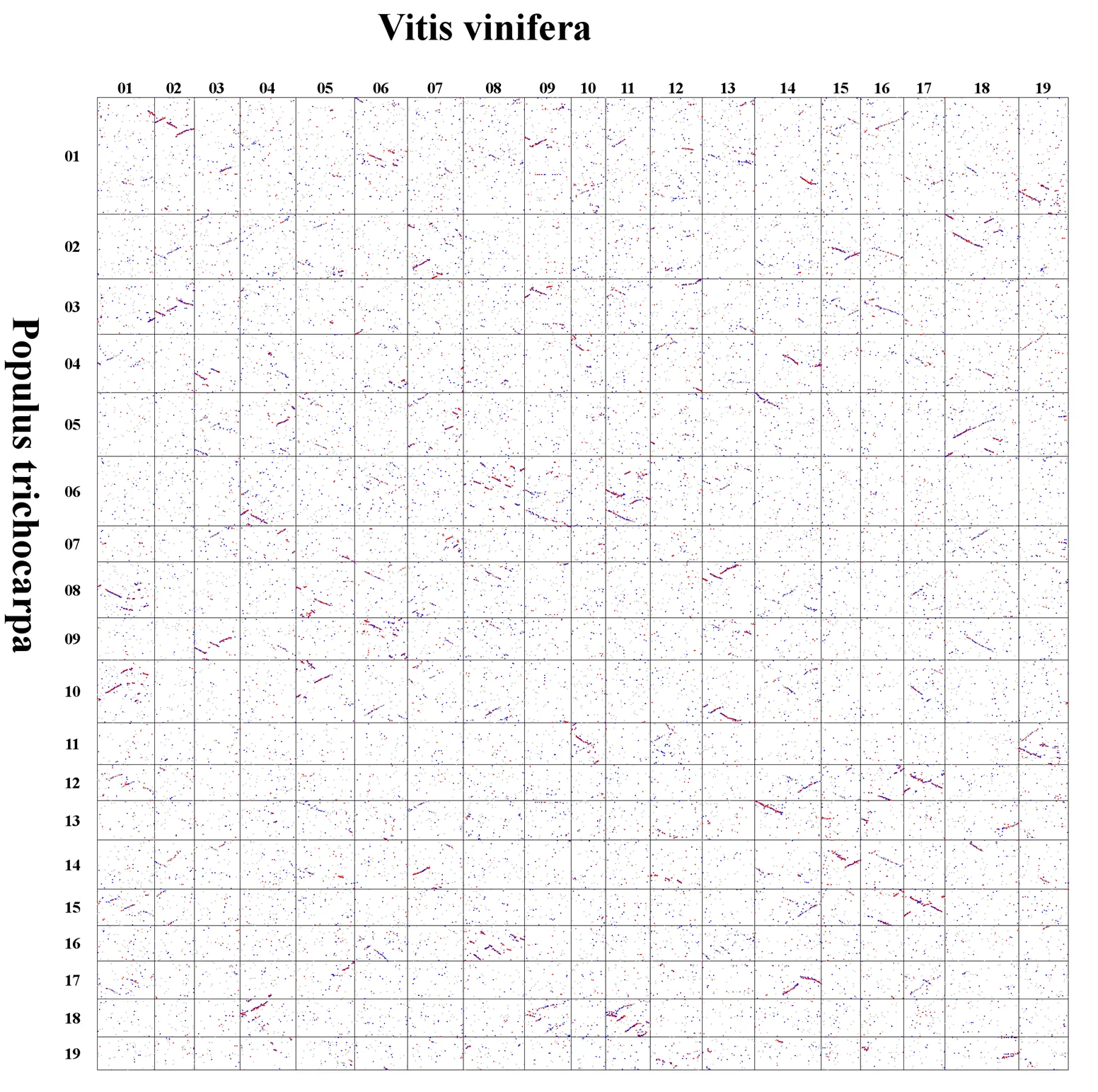
图2 葡萄和毛果杨基因组间同源性点阵图
从图2可见,毛果杨18号染色体与葡萄的4、9、11号染色体具有较完整的同源性,但葡萄的4、9、11号染色体间隔较远。这会给大规模的染色体结构推断过程中带来一定程度上的困难,同时,也难以判断葡萄4、9、11号染色体的同源关系。从图3可见,A1、A2、A3分别与毛果杨的4、8、10、12、15、17染色体都具有较完整的同源片段。由于A1、A2、A3是由同一个祖先染色体A三倍乘而来,因此A1、A2、A3具有明显的同源关系。
从图3中可见,其它同一个字母下的3条染色体也具有十分显著的同源关系,这说明对祖先染色体的拼接过程是正确的。
从图3还可见,毛果杨18号染色体与双子叶植物祖先染色体F1、F2、F3的同源关系非常明显,很容易看出毛果杨的18号染色体是由7条染色体中的F号染色体演变而来的。比较图2、图3不难发现,双子叶植物祖先基因组作为参考基因组时,获得的同源片段更加紧凑和完整。以双子叶植物祖先基因组作为参考基因组得到的同源片段的完整性远远优于以葡萄作为参考基因组的情况。
双子叶共同祖先7条染色体加倍后,应该有21条祖先染色体,而葡萄仅仅有19条染色体,这说明双子叶植物共同祖先演变成葡萄过程中,染色体发生了一定程度的融合。由于葡萄与双子叶植物共同祖先存在较大的差异,因此,在比较基因组学研究中,用葡萄作为外类群参考不如用本文拼接的双子叶植物21条祖先染色体作基因参考组的效果好。
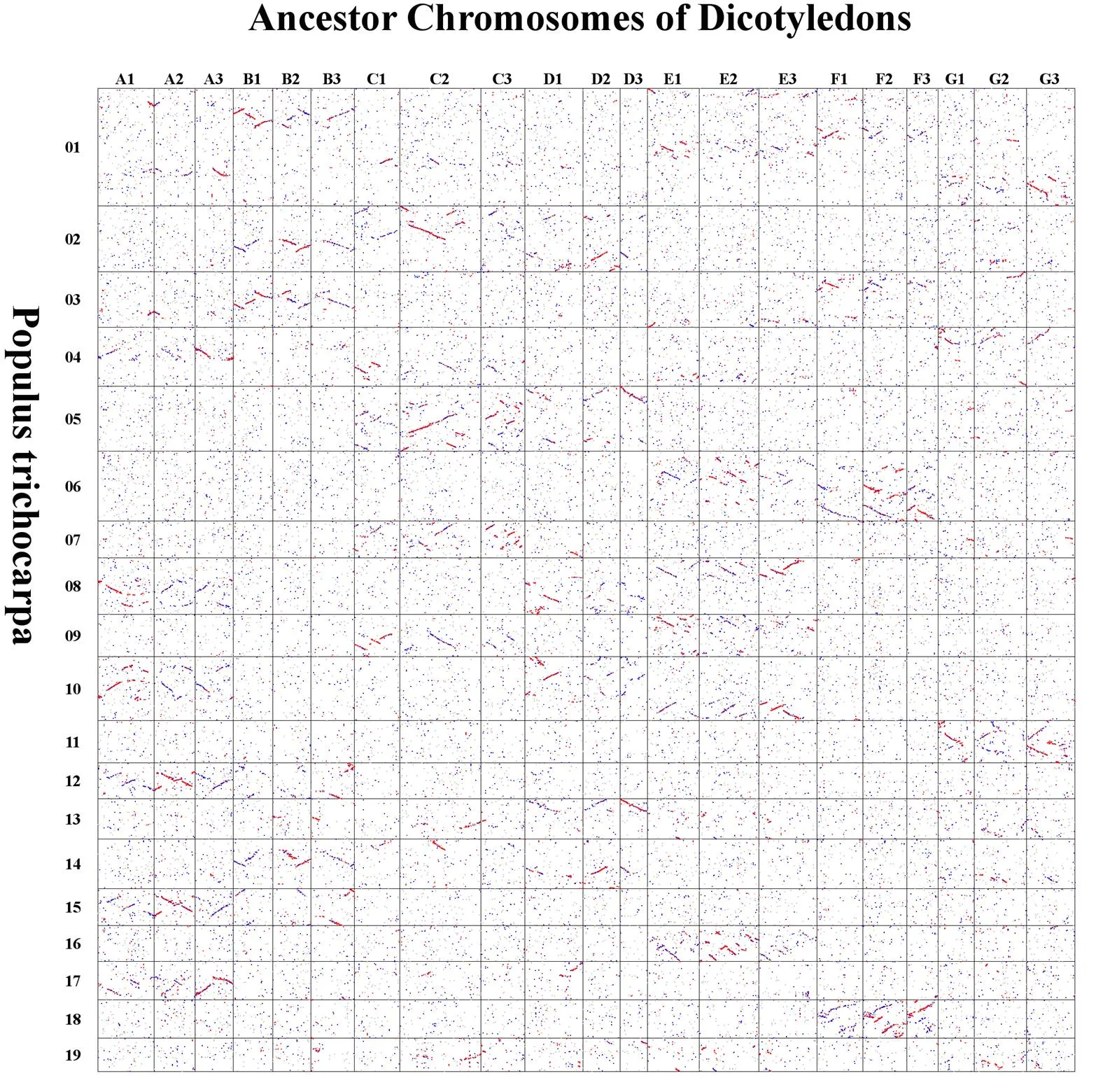
图3 双子叶植物祖先和毛果杨基因组间同源性点阵图
3 结论
按照文献方法拼接的双子叶植物共同祖先基因组是合理的,相对于葡萄基因组,染色体结构更加接近于目前的双子叶植物染色体结构。选取本文提供的双子叶植物共同祖先基因组序列作为参照,可降低可视化同源结构点阵图中同源片段的散乱程度,为现代基因组重构出祖先染色体核型进化过程提供极大便利。
[1] Wang X, Guo H, Wang J, et al. Comparative genomic de-convolution of the cotton genome revealed a decaploid ancestor and widespread chromosomal frac- tionation[J]. New Phytologist, 2016, 209(3): 1252- 1263.
[2] Wang X, Jin D, Wang Z, et al. Telomere-centric genome repatterning determines recurring chromosome number reductions during the evolution of eukaryotes[J]. New Phytologist, 2015, 205(1): 378-389.
[3] Jaillon O, Aury J M, Noel B, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla[J]. Nature, 2007, 449(7161): 463.
[4] Jiao Y, Wickett N J, Ayyampalayam S, et al. Ancestral polyploidy in seed plants and angiosperms[J]. Nature, 2011, 473(7345): 97-100.
[5] Liu Y, Wang J, Ge W, et al. Two Highly Similar Poplar Paleo-subgenomes Suggest an Autotetraploid Ancestor of Salicaceae Plants[J]. Frontiers in Plant Science, 2017, 8(12): 571.
[6] Tuskan G A, Difazio S, Jansson S, et al. The genome of black cottonwood, Populus trichocarpa (Torr & Gray) [J]. Science, 2006, 313(5793): 1596-1604.
[7] Wang X, Shi X, Hao B, et al. Duplication and DNA segmental loss in the rice genome: implications for diploidization[J]. New Phytologist, 2005, 165(3): 937.
[8] Wang J P, Yu J G, Li J, et al. Two Likely Auto- Tetraploidization Events Shaped Kiwifruit Genome and Contributed to Establishment of the Actinidiaceae Family[J]. iScience, 2018,10(7):230-240.
[9] Wang X, Tang H, Bowers J E, et al. Comparative inference of illegitimate recombination between rice and sorghum duplicated genes produced by polyploi- dization[J]. Genome Research, 2009, 19(6): 1026.
[10] Eric S D, Nicholas T K D D, Theophilus K A. Bioinformatics with basic local alignment search tool (BLAST) and fast alignment (FASTA)[J]. Journal of Bioinformatics & Sequence Analysis, 2014, 6(1): 1-6.
[11] Wang X, Shi X, Li Z, et al. Statistical inference of chromosomal homology based on gene colinearity and applications to Arabidopsis and rice[J]. BMC Bioinfor- matics, 2006, 7(1): 1-13.
Application of Ancestor Chromosomes of Dicotyledons in Comparative Genomics
ZHAO Yu-hao, WANG Jin-peng, WANG Xi-yin
(College of Life Sciences, North China University of Technology, Tangshan 063210, China)
Based on the grape genome data and the inference process of seven ancestral chromosomes of dicotyledons, the protein sequences of the triploid common ancestor (21 ancestral chromosomes) of dicotyledons were reconstructed and spliced, and the spliced common ancestral genome data of dicotyledons were used as reference species in comparative genomics with dicotyledons. The results showed that the common ancestors of the spliced dicotyledons as the reference species were more conducive to the comparative genomics study in dicotyledons.
ancestor chromosome; comparative genomics; grape genome; reference species
Q941.2
A
1009-9115(2019)06-0051-04
10.3969/j.issn.1009-9115.2019.06.013
2019-05-0
2019-05-15
赵毓昊(1993-),男,河北唐山人,硕士研究生,研究方向为比较基因组学。
(责任编辑、校对:李春香)
猜你喜欢
基层中医药(2022年4期)2022-07-22
故事作文·低年级(2022年7期)2022-07-18
科学大众(2021年21期)2022-01-18
汉字汉语研究(2021年2期)2021-08-30
学苑创造·A版(2021年6期)2021-08-10
今日农业(2020年19期)2020-12-14
科学(2020年2期)2020-08-24
科学(2020年2期)2020-08-24
汉字汉语研究(2019年2期)2019-08-27
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
