
Brain-derived neurotrophic factor mediates macrophage migration inhibitory factor to protect neurons against oxygen-glucose deprivation
2020-01-18 06:02SuHwanBaeMiRanYooYeYeongKimInKyungHongMiHeeKimSeungHakLeeDaeYulKim
中国神经再生研究(英文版) 2020年8期
Su Hwan Bae, Mi Ran Yoo, Ye Yeong Kim, In Kyung Hong, Mi Hee Kim, Seung Hak Lee, Dae Yul Kim,
1 Department of Rehabilitation Medicine, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republic of Korea
2 Asan Institute for Life Sciences, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republic of Korea
Abstract Macrophage migration inhibitory factor (MIF) is a chemokine that plays an essential role in immune system function. Previous studies suggested that MIF protects neurons in ischemic conditions. However, few studies are reported on the role of MIF in neurological recovery after ischemic stroke. The purpose of this study is to identify the molecular mechanism of neuroprotection mediated by MIF. Human neuroblastoma cells were incubated in Dulbecco’s modified Eagle’s medium under oxygen-glucose deprivation (OGD) for 4 hours and then returned to normal aerobic environment for reperfusion (OGD/R). 30 ng/mL MIF recombinant (30 ng/mL) or ISO-1 (MIF antagonist; 50 μM) was administered to human neuroblastoma cells. Then cell cultures were assigned to one of four groups: control, OGD/R, OGD/R with MIF, OGD/R with ISO-1. Cell viability was analyzed using WST-1 assay. Expression levels of brain-derived neurotrophic factor (BDNF), microtubule-associated protein 2 (MAP2), Caspase-3, Bcl2, and Bax were detected by western blot assay and immunocytochemistry in each group to measure apoptotic activity. WST-1 assay results revealed that compared to the OGD/R group, cell survival rate was significantly higher in the OGD/R with MIF group and lower in the OGD/R with ISO-1 group. Western blot assay and immunocytochemistry results revealed that expression levels of BDNF, Bcl2, and MAP2 were significantly higher, and expression levels of Caspase-3 and Bax were significantly lower in the MIF group than in the OGD/R group. Expression levels of BDNF, Bcl2, and MAP2 were significantly lower, and expression levels of Caspase-3 and Bax were significantly higher in the ISO-1 group than in the OGD/R group. MIF administration promoted neuronal cell survival and induced high expression levels of BDNF, MAP2, and Bcl2 (anti-apoptosis) and low expression levels of Caspase-3 and Bax (pro-apoptosis) in an OGD/R model. These results suggest that MIF administration is effective for inducing expression of BDNF and leads to neuroprotection of neuronal cells against hypoxic injury.
Key Words: apoptosis; brain-derived neurotrophic factor; hypoxia; in vitro; ischemic stroke; macrophage migration inhibitory factor; nerve regeneration; neuroprotective effect; reperfusion
Introduction
Ischemic stroke is a neurological disorder caused by the attenuation of cerebral blood flow. In recent years, the number of ischemic stroke patients is increasing due to the increase of aging population and the influence of a westernized lifestyle (Jung et al., 2012). Typical symptoms are weakness, sensory dysfunction, spasticity, aphasia, dizziness, gait disturbance, and cognitive dysfunction. Neurological deficits caused by stroke could seriously reduce quality of life (Sturm et al., 2004).
Basic and clinical researchers have conducted extensive investigations to better understand stroke and develop new therapeutic strategies (Li et al., 2017). After cerebral infarction, the infarct core develops necrosis, while the effects in the peripheral area of the infarction (penumbra) may be reversible. As a result, many studies have been conducted on neuroprotective treatments that target the penumbra (Ferrer, 2006).
Reduced blood supply to brain tissue can lead to necrosis of neurons, which can then trigger immune responses, leading to inflammatory cell activation and infiltration. These inflammatory reactions in the penumbra after ischemic stroke can cause additional injuries to the brain tissue. Although there is controversy, many studies have reported that inflammation could aggravate ischemic injury (Allan et al., 2005; Muhammad et al., 2008). Therefore, regulation of these inflammatory reactions could be a potential target for ischemic stroke treatment.
Macrophage migration inhibitory factor (MIF) is a chemokine (Tillmann et al., 2013) that plays an essential role in immune system function. MIF is expressed in a variety of cells, including T cells, monocytes, and endothelial cells (Asare et al., 2013). MIF inhibits the migration of macrophages and is associated with various inflammatory and auto-immune diseases (Lue et al., 2002). Several previous studies have suggested that MIF has a protective function during ischemic events. Indeed, one study reported that plasma MIF levels increase in patients after ischemic stroke (Wang et al., 2009). Another study found that MIF expression increased after incubating human glioblastoma cells in hypoxic conditions and that cell survival increased with the administration of MIF compared to the control group (Zis et al., 2015). Thus, authors suggested that MIF protects neurons in hypoxic conditions.
However, there are few reports on the role of MIF in neurological recovery after ischemic stroke. In a previous study, our team tried to identify exercise induced MIF and brain-derived neurotrophic factor (BDNF) after middle cerebral artery occlusion in a rodent (Chang et al., 2019). The expression levels of MIF and BDNF in the exercise groups were significantly higher than those of the non-exercise groups. Thus, we concluded that MIF expression increased by exercise may play a critical role in mediating neuronal regeneration after stroke. Considering these previous studies, we hypothesized that the molecular mechanism of neurological recovery during rehabilitation in ischemic stroke patients is mediated by the MIF action.
Therefore, the purpose of this in vitro study was to identify if MIF is associated with neuroprotection and to identify the molecular mechanism of neuroprotection mediated by MIF in human neuroblastoma cells using an oxygen-glucose deprivation and reperfusion (OGD/R) model.
Materials and Methods
Cell culture
We used human neuroblastoma cell line SH-SY5Y (Cat# CRL-2266, American type culture collection, VA, USA, Research resource identifier (RRID): CVCL_0019) for this study (Figure 1A). Since a commercialized cell line was used, it was confirmed that approval of the ethics committee was not necessary. Cultures were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Biowest, Nuaillé, France), 100 U/mL penicillin, and 100 μg/mL streptomycin (Pen strep, Gibco). Cells were maintained at 37°C in a humidified incubator containing 5% CO2.
Oxygen-glucose deprivation and reperfusion (OGD/R) procedure
Figure 1Bshows a schematic diagram of experimental process. We established an in vitro model of cerebral ischemia by subjecting cells to oxygen-glucose deprivation (OGD). For cells undergoing OGD, cultures were transferred to a multi-gas incubator containing a gas mixture with 1% O2. The medium was replaced with a pre-warmed (37°C) glucose-free DMEM. For removing dissolved oxygen, the solution was bubbled with an anaerobic gas mixture (95% N2, 5% CO2) for 1 hour. When dissolved oxygen was less than 0.1 PPM (mg/L) using a dissolved oxygen measuring kit (CHEMetsR kit, K-7501, CHEMetrics Inc, Midland, VA, USA), cell cultures subjected to OGD were incubated in glucose-free DMEM for 4 hours in a multi-gas incubator and then incubated under normoxic conditions in culture medium containing 10% FBS and glucose for additional 24 hours for reperfusion (OGD/R). We administered 30 ng/mL MIF recombinant (concentrations were selected as per Moon et al., 2012) (Cat# ab7207, Abcam, Cambridge, England, RRID:AB_305760) or 50 μM of the MIF antagonist ISO-1 ((S,R)-3-(4-Hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid, methyl ester, MIF antagonist, Merck, Kenilworth, NJ, USA) after 4 hours of OGD and cultures were incubated under normoxic conditions containing 10% FBS and glucose for additional 24 hours. Cultures in the control group were maintained in high glucose containing DMEM under normoxic conditions for 28 hours. To compare cell viability between different treatments, we assigned cell cultures to one of four groups: control, OGD/R, OGD/R with MIF (MIF), OGD/R with ISO-1 (ISO-1).
WST-1 assay
Cell viability was evaluated using the WST-1 assay. Cells (2.0 × 104cells/well) were grown in 96-well plates (Cat#30096, SPL life Sciences, Pocheon, Korea). After exposure to OGD/R, cells were washed with serum-free DMEM, and 10 μL cell proliferation reagent WST-1 (Roche, Cat. No. 05015944001, Switzerland) was added to each 100 μL/well serum-free DMEM (1:10 dilution), followed by incubation for 1 hour at 37°C. For background measurements, 10 μL WST-1 was added to 100 μL serum-free DMEM. Finally, the absorbance was measured at 440 nm using a microplate reader (REF 16039400, Tecan, Männedorf, Switzerland). The background results were subtracted from each group and each experimental group was compared to the control group.
Western blot analysis
For evaluating the neuroprote ctive effect between groups, we compared gene expression of BDNF (both pro-BDNF and mature BDNF) and microtubule associated protein 2 (MAP2) by western blot analysis. We also analyzed Bcl2 associated X protein (Bax), B-cell lymphoma 2 (Bcl2) and Caspase-3 expression levels to compare apoptotic activity.
Cell pellets were collected using centrifuge and washed to remove supernatant. At 20 minutes after adding 400 μL protein extraction solution (Proprep, iNtRON Biotech, Seongnam, Korea), cell lysates were centrifuged at 16,000 r/min for 20 minutes and the supernatant was collected. Using Bradford assay, the amount of protein obtained from the experiments was quantified. Standard curve was obtained using the absorbance of 2 mg/mL bovine serum albumin (BSA) and the concentration of protein corresponding to the absorbance was calculated. Based on these protein concentrations, each sample was prepared to 1 μg/μL and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDSPAGE). After electrophoresis, proteins were transferred to a nitrocellulose membrane at 2.5 A, 25 V for 13 minutes using a Bio-Rad system (Bio-Rad, Hercules, CA, USA). Tris-buffered saline (TBS) (0.2 M Tris-HCl, 1.37 M NaCl) was diluted in distilled water to a concentration of 1:9 and Tween (TWEEN 20, Sigma, St. Louis, MO, USA) was added up to 0.001%. Tris-buffered saline with Tween (TBS-T) was used to wash solution of the primary and secondary antibodies. The membranes were blocked in 5% BSA/TBS-T for 1 hour. The primary antibodies for Bcl-2 (Cat# ab7973, Abcam, Cambridge, England, RRID:AB_306187), pro-BDNF (Cat# ab72439, Abcam, RRID:AB_1267795), mature BDNF (Cat# ab205067, Abcam), MAP2 (Cat# MA5-12826, Thermo Fisher Scientific, Waltham, MA, USA, RRID:AB_10976831), Caspase-3 (Cat# ab4051, Abcam, RRID:AB_304243) and Bax (Cat# ab53154, Abcam, RRID:AB_867795) were diluted in TBS-T to a concentration of 1:1000 overnight at 4°C and 10 mL per membrane was used. Horseradish peroxidase (HRP) conjugated secondary antibodies (mouse antibody against MAP2 and mature BDNF, mouse IgG antibody, GeneTex, Irvine, CA, USA; rabbit antibody against immature BDNF, Caspase-3, Bcl2, and Bax, goat anti-rabbit IgG, Enzo Life Science, New York, NY, USA) were diluted in TBS-T to a concentration of 1:5000.
Incubation was performed with HRP-conjugated secondary antibodies for 1 hour at room temperature and washed three times with TBS-T. The resultant protein bands were visualized by enhanced chemiluminescence (ECL) western blotting detection reagents according to the manufacturer’s instructions (Thermo Fisher Scientific). β-Actin was used as a loading control.
We quantified the results of western blot assay using Image Studio Lite ver. 5.2 (LI-COR Inc., Lincoln, NE, USA). To calculate protein expression from the same amounts of cells in each group, the band results of each group were divided by the band results of the loading control, β-actin and shown as a ratio of each group relative to the control group.
Immunocytochemistry
For evaluating cellular responses between groups, we analyzed the differences in BDNF (mature BDNF), MAP2, Bcl2, Bax and Caspase-3 protein expression levels with immunocytochemistry. Cells (1 × 105cells/well) were seeded onto a cover slip pre-coated with 2% gelatin solution (Sigma). Then cells were fixed with 4% paraformaldehyde (PFA) for 10 minutes at room temperature and washed three times with phosphate buffered saline (PBS). After permeabilization with 0.1% Triton X-100 (Sigma) in tris phosphate buffered saline (tPBS) for 10 minutes, cells were blocked in 5% normal goat serum (NGS)/5% BSA/0.1% tPBS for 30 minutes and bound with a primary antibody diluted in 5% BSA/0.1% tPBS overnight at 4°C. We used Bcl-2 (Cat# ab7973, RRID:AB_306187; Abcam), mature BDNF (ab205067; Abcam), MAP2 (Cat# MA5-12826, RRID:AB_10976831; Thermo Fisher Scientific), Bax (Cat# ab53154, RRID:AB_867795; Abcam), and Caspase-3 (Cat# ab4051, RRID:AB_304243; Abcam) as primary antibodies. After washing with PBS three times for 5 minutes each, cells were bound with a secondary antibody diluted in 5% BSA/0.1% tPBS for 90 minutes. We used donkey anti-rabbit IgG H&L (Alexa Fluor 488) (Cat# ab150073, RRID: AB_2636877; Abcam), goat anti-mouse IgG H&L (Alexa Fluor 568) (Cat# ab175473; Abcam) and goat anti-rabbit IgG H&L (Alexa Fluor 488) (Cat# ab150077, RRID:AB_2630356; Abcam) as secondary antibodies at room temperature for 1 hour. After washing with PBS three times for 5 minutes each, cells were mounted with Fluoromount-G and cultured with 10 μL 4′,6-diamidino-2-phenylindole (DAPI) (Cat# 00-4959-52, Invitrogen, Carlsbad, CA, USA) overnight. After overnight staining, we inspected the prepared samples using confocal microscopy (confocal laser scanning microscope; LSM 710, Carl Zeiss, Jena, Germany).
The results of immunocytochemistry were quantified using Image J ver. 1.48 (Carl Zeiss). Cells were observed under a confocal microscope and expressed as the average of the intensity of fluorescence observed in 3-5 or more microscopic fields. The intensity of fluorescence for each cell was divided by DAPI intensity. All samples were washed with PBS multiple times before DAPI staining. Therefore, we assumed that dead cells were washed away, leaving only live cells in the sample. Because DAPI stains the nucleus of a cell, it reflects the number of living cells in the field. Immunoreactivity was calculated as the intensity per cell.
Statistical analysis
Quantifications were performed with three or more independent experiments. Five independent experiments in the WST-1 assay, five or more independent experiments in western blot assay and immunocytochemistry were performed. Data are expressed as the mean ± SD. All statistical analyses were performed using SPSS 18.0 software (SPSS, Chicago, IL, USA). We performed one-way analysis of variance (ANOVA) and post hoc analysis with the Tukey’s honestly significant difference test or Bonferroni methods to determine statistical significance of comparisons. Values of P < 0.05 were considered statistically significant.
Results
WST-1 assay
Figure 2 shows cell survival results from the WST-1 assays. Relative to the OGD/R group, cell survival was higher in the MIF group and lower in the ISO-1 group (OGD/R, 0.81 ± 0.12; MIF, 0.87 ± 0.07; ISO-1, 0.72 ± 0.06, n = 5, number of independent cell culture experiments). These results were statistically significant (P = 0.03).
Western blot analysis
Figure 3 shows the expression levels of various proteins associated with neuronal regeneration and apoptosis from the western blot analysis. All data were expressed as ratios relative to the control group. The expression levels of immature and mature BDNF were significantly higher in the MIF group and lower in the ISO-1 group than those in the OGD/R group (Figure 3A; P < 0.05 for immature BDNF, P < 0.01 for mature BDNF). Compared with the OGD/R group, the expression levels of Bcl2 (anti-apoptotic marker) and MAP2 (neuronal marker) were significantly higher in the MIF group and lower in the ISO-1 group (Figure 3A; P < 0.01 for Bcl2, P < 0.05 for MAP2). Expression levels of Caspase-3 and Bax (apoptotic marker) were significantly lower in the MIF group and higher in the ISO-1 group than those in the OGD/R group (Figure 3B; P < 0.01 for Caspase-3, P = 0.02 for Bax).

Figure 2 Cell survival assessed by the water-soluble tetrazolium salts-1 assay relative to the control group.

Figure 3 Western blot results of neuroblastoma cell protein expression under control, OGD/R conditions, and OGD/R conditions treated with exogenous MIF or MIF antagonist ISO-1.
Immunocytochemistry
Figure 4 shows the fluorescence immunocytochemistry staining of neuroblastoma cells for various proteins associated with neuronal regeneration and apoptosis. To present protein expression per cell, all data were expressed as ratios for each fluorescence intensity relative to DAPI intensity, which correspond to the relative number of live cells. The expression level of mature BDNF was significantly higher in the MIF group and lower in the ISO-1 group than that in the OGD/R groups (Figure 4A and B; P < 0.01). Similarly, the expression level of MAP2 (neuronal marker) was significantly higher in the MIF group and lower in the ISO-1 group than that in the OGD/R group (Figure 4A and B; P < 0.01). The mean expression level of Bcl2 (anti-apoptotic marker) tended to be higher in the MIF group than in the ISO-1 group (Figure 4A and B, P = 0.03 but not significant in the post-hoc analysis). The expression levels of Caspase-3 and Bax (apoptotic marker) were significantly lower in the MIF group and higher in the ISO-1 group than those in the OGD/R group (Figure 4C and D; P < 0.01 for both Caspase-3 and Bax).
Discussion
Based on the findings of our previous in vivo study, this in vitro study investigated how MIF affects neuronal cells in an ischemia and reperfusion environment. Our results suggest that MIF administration promotes cell survival and anti-apoptotic pathways in ischemic neuroblastoma cells.
In the WST-1 assay, cell survival in the MIF group was higher than that in the OGD/R group while it in the ISO-1 group was lower than that in the OGD/R group. These results imply that MIF administration has a direct neuroprotective effect. MIF is a multipotent cytokine and acts by interacting with various inflammatory cells including T lymphocytes (Kasama et al., 2010). Another study suggested that MIF can promote the proliferation of neural stem cells and progenitor cells in vitro (Ohta et al., 2016).
Our results also suggested that MIF directly affected neuronal cells without involvement of inflammatory cells, leading to intracellular molecular responses that may be important in neuroprotective function. Also, lower cell survival in the ISO-1 group compared to the OGD/R group was attributable to the inhibition of endogenous MIF by ISO-1 action. Furthermore, the trends of cell survival between four groups were consistent with the expression levels of BDNF and proteins involved in apoptosis.
BDNF is an essential modulator of synaptic plasticity in the central nervous system, supporting neuronal survival and promoting growth and differentiation of new neurons (Hu et al., 2019). BDNF is initially produced in an immature form, pro-BDNF, and then degraded into a mature BDNF. Pro-BDNF binds to the pan-neurotrophin receptor p75NTR, and mature form BDNF binds to the tyrosine kinase TrkB (Chao and Bothwell, 2002; Ibanez, 2002). Pro-BDNF is associated with long term depression and mature BDNF is involved in the early stage of long-term potentiation (Korte et al., 1995; Woo et al., 2005). One study reported that longterm exercise is associated with an increase in MIF, leading to increases in BDNF expression level, indicating an antidepressant effect of exercise (Moon et al., 2012). Another study suggested that serum MIF levels are increased in intracerebral hemorrhage patients and that serum MIF could be a potential prognostic biomarker (Lin et al., 2017). However, our findings suggest that MIF administration increases the expression of not only pro-BDNF but also mature BDNF. Thus, MIF administration may promote neuronal regeneration or neuroprotection by inducing higher BDNF levels after an ischemic event.
The impact of MIF on ischemic stroke remains controversial. Previous studies suggested that MIF has a protective effect on brain ischemia but other studies suggested that MIF has a harmful effect by promoting inflammation in the brain. One study reported that MIF could increase the inflammation under pathologic conditions, which may aggravate additional brain injury in stroke patients (Inacio et al., 2011b). In contrast, Inacio et al. (2011a) reported that MIF did not affect the inflammatory process after stroke. Zhang et al. (2014) reported that MIF decreased Caspase-3 activity and protected neurons from ischemia/reperfusion induced apoptosis. Nishio et al. (2002) revealed that MIF promoted peripheral nerve regeneration and prevented the apoptosis of Schwann cells (2002). In our study, the results suggested that MIF promoted not only mature BDNF formation, but also increased the expression of anti-apoptotic marker Bcl2 and decreased the expression of apoptotic marker Caspase-3 and Bax in human neuroblastoma cells. Also, MAP2, a neuronal marker, increased in the MIF group, which could be interpreted as an increase in the number of nerve cells. The results of western blot analysis were consistent with those of immunocytochemistry. The expression levels of mature BDNF, Bcl2, and MAP2 in the MIF group were higher and the expression levels of Caspase-3 and Bax in the MIF group were lower than those in the OGD/R group. The opposite results were obtained in the ISO-1 (MIF antagonist) group. Cell morphology was generally good in the OGD/R cells treated with MIF, while apoptosis was frequently observed in OGD/R cells treated with ISO-1. These results imply that MIF administration may be effective for neuroprotection in ischemia and reperfusion injury.
In ischemic stroke treatment, rehabilitation is becoming increasingly important for recovery. However, the mechanism of rehabilitation for neurological recovery is unclear. Our previous in vivo findings (Chang et al., 2019) and current in vitro results suggest one possible mechanism of neurological recovery from rehabilitation in stroke patients; exercise or rehabilitation may induce MIF activity that promotes neuroprotection and neuronal regeneration. Furthermore, MIF administration may help restore neuronal injury, and could be a novel therapeutic modality for neuroprotection in ischemic stroke patients.
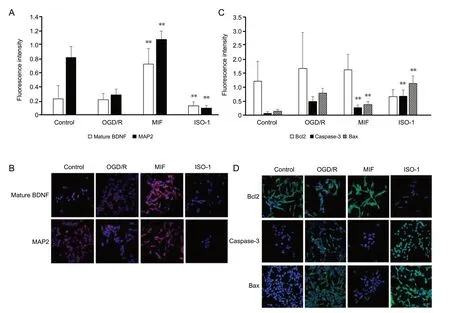
Figure 4 ICC findings of neuroblastoma cells under control, OGD/R conditions, and OGD/R conditions treated with exogenous MIF or MIF antagonist ISO-1.
There are several limitations for this study. First, we did not investigate the changes in the degree of neuronal cell injury and response according to duration of hypoxia and dose of MIF. Based on previous studies, we determined the dose of MIF (30 ng/mL) and duration of hypoxia (4 hours). We expect to evaluate the changes of neurons according to dose of MIF and duration of hypoxia in a future study. Second, we used human neuroblastoma cells instead of normal human nerve cells in this study. In general, cancer cells can be more resistant to hypoxic environments than normal cells. Thus, the responses observed in the OGD/R model may differ from those of normal cells. A further study identifying changes in normal neuronal cells for hypoxic environment with MIF administration will be necessary. Lastly, we did not evaluate the role of MIF in the tissue or organ level. MIF is known to play a role through interaction with various inflammatory cells. However, in our study, we evaluated only intracellular responses directly induced by MIF and ISO-1 administration. Further in vivo studies will be necessary to evaluate the mechanisms of MIF.
In conclusion, this study revealed that MIF administration promoted neuronal cell survival, induced the expression of mature BDNF, MAP2 and Bcl2, and decreased the expression of Caspase-3 and Bax in OGD/R model. These results imply that MIF administration is effective for neuroprotection in hypoxic neuronal cells. Thus, MIF could be a novel therapeutic modality for neuroprotection of cerebral ischemia and reperfusion injury.
Author contributions:Conception and design of this study: SHB, IKH, SHL, DYK; data collection: YYK, IKH, MHK, MRY, SHL, DYK; data analysis: SHB; paper writing: SHB, SHL, DYK. All authors approved the final version of this study.
Conflicts of interest:None declared.
Financial support:This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, No. 2016R1A2B4012772 (to DYK). The funding body played no role in the study conception and design, in the critical revision of manuscript for intellectual content, and in the writing of the paper, and in the decision to submit the paper for publication.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Exercise promotes recovery after motoneuron injury via hormonal mechanisms
- Large animal ischemic stroke models: replicating human stroke pathophysiology
- Autophagy and inflammation in ischemic stroke
- Electrical stimulation and denervated muscles after spinal cord injury
- Beneficial effects of saffron (Crocus sativus L.) in ocular pathologies, particularly neurodegenerative retinal diseases
- Toxic tau: structural origins of tau aggregation in Alzheimer’s disease