
Toxic tau: structural origins of tau aggregation in Alzheimer’s disease
2020-01-18 06:02AbdullahAlMamunMdSahabUddinBijoMathewGhulamMdAshraf
中国神经再生研究(英文版) 2020年8期
Abdullah Al Mamun, Md. Sahab Uddin, , Bijo Mathew, Ghulam Md Ashraf
1 Department of Pharmacy, Southeast University, Dhaka, Bangladesh
2 Pharmakon Neuroscience Research Network, Dhaka, Bangladesh
3 Division of Drug Design and Medicinal Chemistry Research Lab, Department of Pharmaceutical Chemistry, Ahalia School of Pharmacy, Palakkad, India
4 King Fahd Medical Research Center, King Abdulaziz University, Jeddah, Saudi Arabia
5 Department of Medical Laboratory Technology, Faculty of Applied Medical Sciences, King Abdulaziz University, Jeddah, Saudi Arabia
Abstract Alzheimer’s disease is characterized by the extracellular accumulation of the amyloid β in the form of amyloid plaques and the intracellular deposition of the microtubule-associated protein tau in the form of neurofibrillary tangles. Most of the Alzheimer’s drugs targeting amyloid β have been failed in clinical trials. Particularly, tau pathology connects greatly in the pathogenesis of Alzheimer’s disease. Tau protein enhances the stabilization of microtubules that leads to the appropriate function of the neuron. Changes in the quantity or the conformation of tau protein could affect its function as a microtubules stabilizer and some of the processes wherein it is involved. The molecular mechanisms leading to the accumulation of tau are principally signified by numerous posttranslational modifications that change its conformation and structural state. Therefore, aberrant phosphorylation, as well as truncation of tau protein, has come into focus as significant mechanisms that make tau protein in a pathological entity. Furthermore, the shape-shifting nature of tau advocates to comprehend the progression of Alzheimer’s disease precisely. In this review, we emphasize the recent studies about the toxic and shape-shifting nature of tau in the pathogenesis of Alzheimer’s disease.
Key Words: Alzheimer’s disease; neurofibrillary tangles; shape-shifting nature of tau; tau aggregation; toxic tau
Introduction
Alzheimer’s disease (AD) is an irreversible progressive disorder that is featured by the most remarkable symptoms including memory loss and impairment of cognition (Kabir et al., 2019b; Uddin et al., 2019c). However, the specific process by which those symptoms progress remains abstruse. The foremost histopathological features of AD are the atypical aggregation of amyloid β (Aβ) and the tau protein (Kabir et al., 2019a; Uddin et al., 2019b). A number of researches recommend that in AD, the tiny oligomeric forms of both tau and Aβ may work synergistically to facilitate synaptic dysfunction. Most importantly, tau pathology plays a pivotal role in the development of AD than Aβ. In recent times, several studies have commenced advocating that the missorting of tau protein between the axon and the dendrites is a prerequisite to intervene with the harmful effects of Aβ (Guerrero-Muñoz et al., 2015; Uddin et al., 2019a).
The misfolding of protein is the preliminary phase in the accumulation process of both tau and Aβ (Uddin et al., 2020; Uddin and Kabir, 2019). There are two essential factors such as post-translational modifications and the formation of disulfide bridges raise the capability of both tau and Aβ proteins to self-assemble into oligomers (Sahara et al., 2007). It is evident that before the fibrils formation tau monomer is initially transformed into an oligomeric state. Moreover, accumulation of tau does not take place spontaneously but the fibril formation is induced by the combination of free fatty acids and polyanionic compounds (King et al., 2002). These different structures vary both in accumulation state and in their toxic actions. This review represents the precise point at which a healthy protein becomes toxic and shape-shifting nature of tau that aggregate and contribute to AD pathogenesis.
Toxic Tau and Alzheimer’s Pathogenesis
Experimental data recommend that tau in prefilamentous forms, precisely oligomers are considered as the neurotoxin. Before the commencement of the medical symptoms, oligomers of tau have been sequestered at very initial phases of the disease. Tau oligomers demonstrate a sphere-shaped morphology by using atomic force microscopy which links with two or more tau molecules, ranging from 6 to 20 nm (Sahara et al., 2008). In fact, these are dynamic structures that turn into β-sheet rich. The study of Himmelstein et al. (2012) exhibited that in AD brain samples, oligomers of tau were found at a 4-fold greater concentration than healthy control samples. Tau is aberrantly phosphorylated at various positions in AD as shown in Figure 1. Conversely, this may not only be a prerequisite for converting tau into oligomers as well as become toxic.
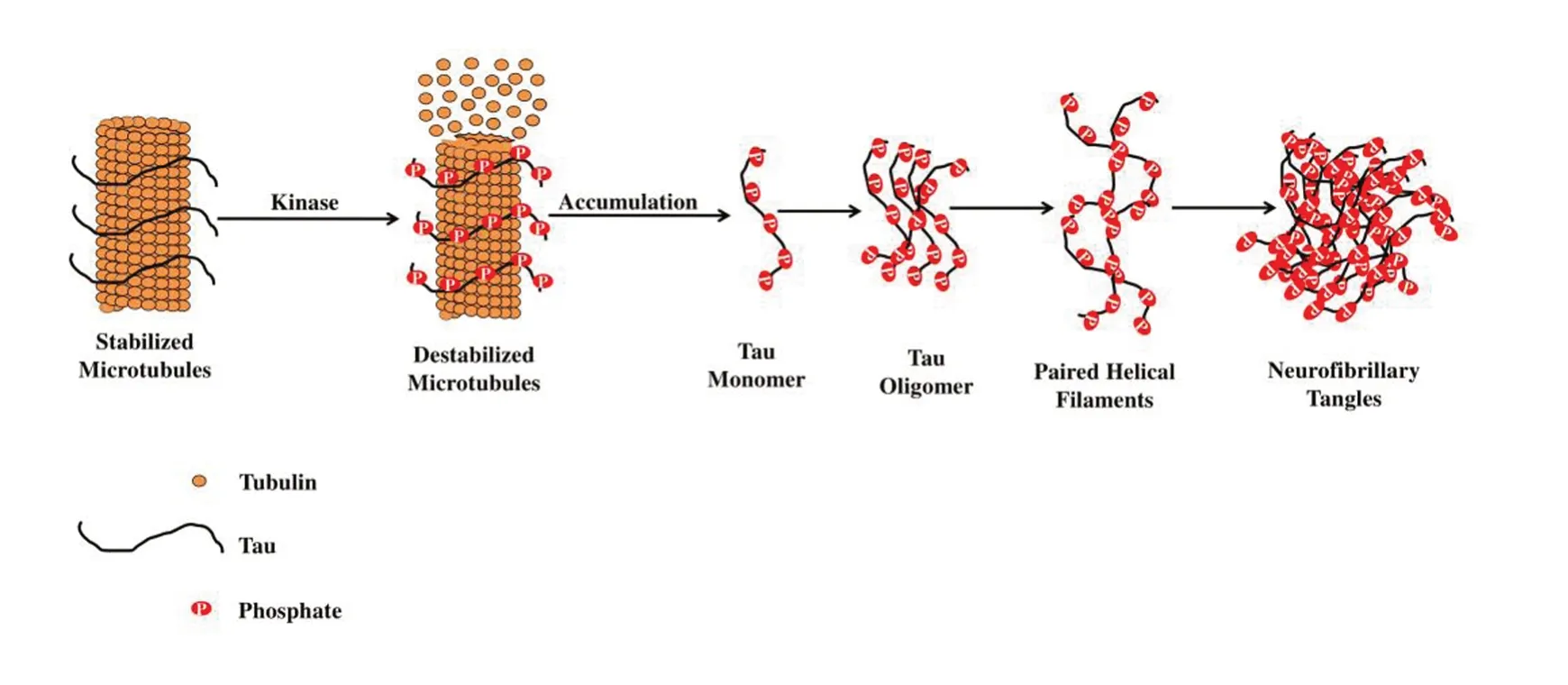
Figure 1 Stabilization of microtubule-associated tau protein is controlled by kinases.
The detrimental accumulation trail inside a cell is assumed to initiate with a seed — a template that can prompt the aggregation of a specified protein. These seeds are believed to be vital in the disseminate of the disease. The study demonstrated that highly sulfated heparan sulfate (HS) aggregates with neurofibrillary tangles in the affected neurons of the AD brain (Snow et al., 1990). In the tau amyloidogenic process, the kinetic constants featuring the development of tau fibrils in the existence or deficiency of heparin agrees with a significant role of HS. Consequently, heparin can interact with two tau molecules yielding a dimer in vitro which is capable of forming fine tiny fibrils (Ramachandran and Udgaonkar, 2011). Conversely, a plethora of heparin can linger the accumulation of tau lag phase, recommending a regulatory role throughout the nucleation phase (Zhu et al., 2010).
Spreading and Propagating of Tau in Alzheimer’s Disease
Some steps such as cellular uptake, templated seeding, secretion and intercellular transfer through synaptic and non-synaptic pathways are anticipated to incorporate for the proliferation of tau pathology by using prion-like mechanisms (Mudher et al., 2017). It is unclear that accumulated tau can be absorbed by cells via precise mechanisms. Ingestion of tau accumulation is done by macropinocytosis (Holmes et al., 2013; Falcon et al., 2015) as well as needs HS proteoglycans (Holmes et al., 2013). After ingestion, tau seeds present in endosomes and must allow contacting with the cytosol to trigger the accumulation of non-accumulated tau.
Two experiments have claimed that in a transgenic mouse line, obvious trans-synaptic movement of tau protein accumulates depend on area-specific gene expression. Tetracycline-controlled gene expression was conducted principally in the entorhinal cortex in both studies that project axons to the hippocampus. In addition, aggregate pathology that was most probably to have derived from the entorhinal neurons was found in the hippocampus in older animals (de Calignon et al., 2012; Liu et al., 2012). The study of Dujardin et al. (2014) using a lentivirus-directed rat model of hippocampal tauopathy showed that wild-type tau is shifted through axons to faraway second-order neurons. Moreover, these investigations powerfully recommended that accumulated tau were passing across synapses, and therefore could possibly elucidate the connection of neural systems in neurodegenerative diseases. Ultimately, current study offerings irrefutable indication that tau proliferates steadily distinctive accumulate conformations, or strains, in cells and mice, as well as that human tauopathies are comprised of disease-related strains (Sanders et al., 2014).
HS can take part both in the tau accumulation processes and its aberrant phosphorylation in the AD brain that may precede or concurrently takes place with its accumulation (Maïza et al., 2018). Apart from this prospective role that HS appears to play in the tau phosphorylation processes as well as accumulation, these polysaccharides have also played an essential role in the proliferation of tau proteopathic particles/seeds between one cell and another cell, an event called as spreading (Holmes et al., 2013). Undeniably, tauopathy is identified in specific brain areas in early AD whereas, as the disease develops, tauopathy seems in other areas (Goedert and Spillantini, 2017).
Deadly Shape-Shifting Nature of Tau in Alzheimer’s Pathology
The postulate is that seeds can alter the typically folded protein into an accumulation of the identical protein, prior to cells release them into the surroundings for adjoining cells to start (Eisenberg and Jucker, 2012). Furthermore, this could be exactly how diseases associated with the tau protein, for instance, AD proliferate from one cell to another. Then, there, the accumulation would travel via the brain using the links from neurons to neurons (Sanders et al., 2014). Although, most of the researchers have thought that they are an aggregate of a given distinct misfolded protein, the identity of the seeds remains vague, hitherto. Mirbaha et al. (2018) recommended that the presence of a stable form of a distinct tau protein that can initiate the accumulation process on its individual.
It is evident that the seed may not only be an aggregate of a misfolded protein but instead be a protein monomer — with a diverse structure. The study of Chirita et al. (2005) anticipated that an alteration in the structure of a tau monomer had played a key role in promoting the progression of accumulation. Another study by Kar et al. (2011) postulated that the accumulation of the protein of huntingtin is connected with a different amyloid disorder called Huntington’s disease, that could primarily begin with a single protein. Nevertheless, according to these studies, the monomer which could start the process of seeding was not separated and studied. Despite strong data interpretation, the majority of the researchers dismissed the principle of monomeric seeds. Additionally, most of the scientific community unwilling to challenge the extensively in-built idea that they are instead an aggregate of a misfolded protein.
Hitherto, tau was characterized by an inherently disordered protein that looked similar to a flexible noodle than a precise, stable, and 3-dimensional protein structure (Schweers et al., 1994). Alternatively, Mirbaha et al. (2018) demonstrated that the tau protein can easily fold into two discrete and properly well-defined structures. Furthermore, one of these shapes is stable, does not simply aggregate, nontoxic; while the other acts as a seed that can aid to transform another innocuous tau monomer into a misfolded tau which will eventually generate toxic accumulations by seeding or self-aggregate. Moreover, tau can alter very slowly from an inactive to the seed-competent structure. It is well-known that tiny molecules can easily bind to an inactive structure of proteins that are susceptible to misfolding, and consequently stop the conformational change which plays a crucial role in the progression of amyloid diseases (Johnson et al., 2012). Although, the accumulation of Aβ and the aggregation of tau are the two central pathological hallmarks of AD, however, Lewy bodies composed of α-synuclein protein are also observed in above 50% of sporadic AD cases investigated (Hamilton, 2000). In a study, Larson et al. (2012) disclosed that toxic, non-fibrillar α-synuclein is considerably raised in AD cases in the absence of Lewy body pathology. Finally, α-synuclein and oligomeric tau interact and co-accumulate in disease (Sengupta et al., 2015) recommends that the two proteins may work in a toxic synergistic mechanism at the synapse in AD brain.
Kinetic Stabilizers to Combat Alzheimer’s Disease
Transthyretin is one of the proteins with two means of folding, and whose toxic structure impairs various parts in the body especially nervous systems. Conversely, drugs are called kinetic stabilizers that can abate the degenerative process by rising the correctly folded structure. To be precise, three placebo-controlled clinical trials revealed that tiny molecules, including the drugs diflunisal and tafamidis, can simply bind to the non-pathogenic form of transthyretin and stabilize it, thus stops the protein from transforming into the structure that starts accumulations and plays an essential role in the development of the degenerative pathologies (Rosenblum et al., 2018). The study has detected structural factors on disease modified tau protein which have played a pivotal controlling role in the development of neurofibrillary pathology in AD. The detected tau factors that are acknowledged by monoclonal antibody DC8E8, considered as a principal for the outline of an active vaccine AADvac1 which went into the phase II clinical trial on AD patients (Novak et al., 2016). Finally, this recommends that it should be likely to approach alike kinetic stabilizers for the tau protein, as well as provide a better treatment option for Alzheimer’s.
Conclusions
It is well-known that the hyperphosphorylated tau is closely connected with neurodegeneration as well as cognitive dysfunction in AD. It has been admitted that aberrant forms of tau protein are directly associated with the commencement of neurodegenerative processes of AD. The current study recognized the concept of shape-shifting which might connect with other proteins that generate toxic accumulates. Therefore, this can support to comprehend clearly the development and progression of neurodegenerative events. Remarkably, the structures that tau forms could be classified as either ‘good’ or ‘bad’ might also aid to advance novel therapies for AD. Medicine can be formulated whether to eradicate the bad form of tau from the brain or to stabilize the good form of tau. Furthermore, if the shape-shift could be diagnosed initially in patients, it might permit therapies for AD prior to patients have developed any evident symptoms.
Acknowledgments:The authors concede the support by the Pharmakon Neuroscience Research Network, Dhaka, Bangladesh.
Author contributions:This work was carried out in collaboration between all authors. All authors read and approved the final submitted version of the manuscript.
Conflicts of interest:The authors proclaim no conficts of interest.
Financial support:None.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Exercise promotes recovery after motoneuron injury via hormonal mechanisms
- Large animal ischemic stroke models: replicating human stroke pathophysiology
- Autophagy and inflammation in ischemic stroke
- Electrical stimulation and denervated muscles after spinal cord injury
- Beneficial effects of saffron (Crocus sativus L.) in ocular pathologies, particularly neurodegenerative retinal diseases
- Modification of tubular chitosan-based peripheral nerve implants: applications for simple or more complex approaches