
NSD2在t(4;14)多发性骨髓瘤中的致病机制研究进展
2020-01-17 02:23李佳慧徐文琦吴飞珍
浙江临床医学 2019年12期
李佳慧 徐文琦 吴飞珍
多发性骨髓瘤(multiple myeloma,MM)是一种浆细胞肿瘤,也是第二大最常见的恶性血液肿瘤,发病率约占所有血液病的13%,占所有肿瘤的1%[1],每年全世界约有130,000 新增MM 病例[2]。其临床症状表现为浆细胞定植骨髓微环境、产生单克隆抗体,引发贫血、溶骨性骨损伤、肾功能衰竭,以及免疫缺陷等。几乎所有的MM 患者均会经历一个恶性前阶段,即具不明意义的单克隆丙种球蛋白病(monoclonal gammopathy of undetermined significance,MGUS), 随后发展为恶性前无症状多发性骨髓瘤(smoldering multiple myeloma,SMM),最后是有症状的恶性MM(multiple myeloma,MM)。MM 目前仍是一种高病死率的不可治愈的恶性肿瘤。
大量对临床患者的遗传学研究显示,MM 具有较高的异质性和个体间差异[3-4],根据初步的核型分析可以将MM 分为两类:超二倍体型(hyperdiploid MM)和非超二倍体型(non-hyperdiploid MM),前者是指患者通常含有一至多个奇数号染色体的三体现象,如3,5,7,9,11,15,19,21 号染色体,所以染色体数目会在48~75 之间变化,而后者通常是包含涉及免疫球蛋白重链基因位点的染色体易位[5],后者发病率更高,约占70%,临床显示非超二倍体型的MM 预后更差[6]。t(4;4)型MM 是指在MM 患者中存在4 号染色体p16 区域与14 号染色体q32 区域的易位,这是MM中第二大常见的一种染色体易位变异,发病率约占所有MM 的15%~20%[7],这种易位会导致两个基因的过表达—FGFR3 和NSD2[8]。但临床数据研究显示只有70%左右的t(4;14)患者会发生FGFR3 的过表达,而所有的都会发生NSD2 的过表达[9],说明NSD2在t(4;14)MM 病程中发挥更重要和更普遍的作用,因此研究清楚NSD2 在其中的作用机制对于t(4;14)MM 的治疗具有重要意义。
1 组蛋白甲基转移酶NSD2的结构与功能
Nuclear receptor binding SET domain 2 protein(NSD2),也叫做multiple myeloma SET domain protein(MMSET) 或 者Wolf-Hirschchorn syndrome candidate 1(WHSC1),是一个组蛋白赖氨酸甲基转移酶,属于NSD 家族,该家族成员还包括NSD1 和NSD3[10]。NSD2 基因位于4 号染色体p16.3 区域,约120kb,包含24 个外显子,存在两种可变剪切转录本和三种蛋白亚型,Ⅰ型转录本编码的NSD2 有647 个氨基酸(short-type),Ⅱ型转录本编码的NSD2 有1365 个氨基酸(long-type),此外,还存在一个从NSD2 内部内含子开始转录产生的蛋白亚型response element Ⅱ binding protein(RE-ⅡBP),三种蛋白亚型的结构如图1 所示,NSD2 是一个多结构域蛋白,Ⅱ型含有1 个SET甲基转移酶活性结构域,2 个PWWP 结构域,一个HMG 结构域和4 个PHD 锌指结构域,后三种结构域对NSD2 与其他蛋白的结合、组蛋白修饰的识别以及染色质定位有重要作用;I 型NSD2 不包含SET 催化结构域,但仍可能通过与其他蛋白的相互作用发挥染色质结构调节功能;RE-Ⅱ BP 与Ⅰ型NSD2 碳端部分重叠,不包含氮端核定位序列,可能主要在细胞质中发挥甲基转移酶活性,甲基化非组蛋白底物。
关于NSD2 的催化底物此前的研究比较混乱,最早的研究报道称NSD2 是H4K20 甲基转移酶[11],此后一系列研究又发现NSD2 还可以催化H3K4me2、H3K9me2、H3K27me3、H3K36me2、H3K36me3 和H4K20me2 等组蛋白修饰[12],这些不一致的结果可能主要是由采用的实验方法和材料差异导致的,比如利用合成的带某种修饰的肽段或组装的核小体或体内纯化的核小体进行体外pull-down 实验时,结果会有差异,最新的研究确认NSD2 主要发挥K3K36me2 甲基转移酶活性[13]。
NSD2 在个体发育中有着重要作用,NSD2 单倍体剂量不足会导致Wolf-Hirschchorn 综合征,临床症状表现为发育迟缓、智力低下、先天性心脏病以及抗体产生缺陷等[14]。NSD2与癌症发生也密切相关,所有(t4;14)MM 中都存在NSD2 的过表达,在胃癌、结直肠癌、肺癌、皮肤癌中也存在过表达[15],且其过表达与膀胱癌、乳腺癌、前列腺癌、神经母细胞瘤的预后差相关[16],此外,有研究发现在多种儿童急性淋巴白血病细胞系中存在E1099K 的突变,该突变导致NSD2 增强的甲基转移酶活性[17]。
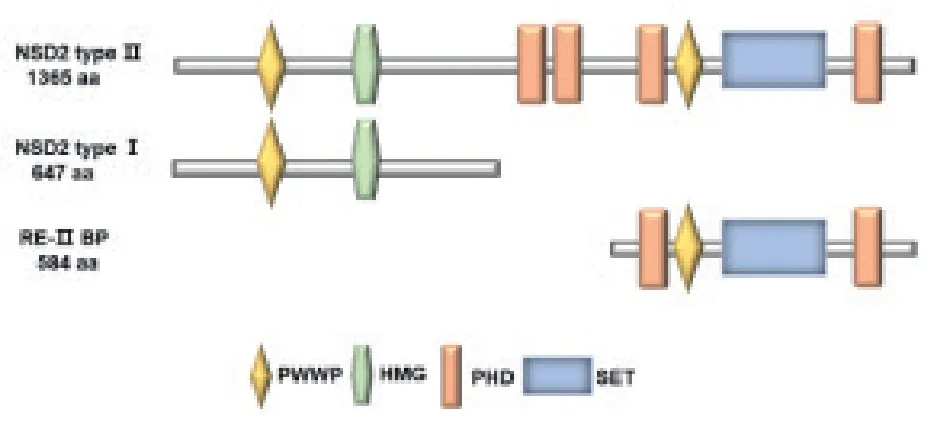
图1 不同NSD2蛋白亚型结构
2 NSD2在t(4;14)MM中的致病机制
NSD2 是一种表观修饰酶,通过组蛋白甲基化修饰可以直接调控基因表达。NSD2 催化产生H3K36me2,H3K36me2 可调控基因表达。NSD2 可与其它组蛋白修饰酶、microRNA,以及其他蛋白相互作用。下面从四方面介绍NSD2 在MM 中的致病机制。
2.1 NSD2 催 化H3K36me2 调 控 基 因 表 达 t(4;14)MM 中NSD2 的过表达会引发全基因组水平的H3K36me2 水平的升高[18],基因体内H3K36me2 是基因转录激活的一个标记,因此许多下游与细胞周期、EMT、细胞迁移以及凋亡有关的原癌基因会被上调,促进MM 的发生发展。Jonathan D. Licht 研究组2011年率先在全基因组水平研究了NSD2 过表达对MM 细胞系KMS11 基因表达的影响,发现H3K36me2 全基因组水平的升高与基因转录激活相关,通过比较NSD2过表达、敲低、敲除以及回补的四种不同细胞系的基因表达谱鉴定到一组NSD2 的靶基因,KEGG 富集分析发现这些基因主要与细胞死亡、细胞周期、DNA 修复以及细胞粘附有关[18]。同年Or Gozani 研究组发表的工作也发现类似现象,NSD2 通过其组蛋白甲基转移酶活性引起KMS11 细胞系全基因组H3K26me2 水平的升高,而基因表达水平与H3K36me2 水平呈正相关,定量PCR 验证了TGFA,PAK1,MET,RRAS2 等基因直接受NSD2 的调控,H3K36me2 水平的升高导致了原癌基因的上调[13],因此,在KMS11 细胞系中通过shRNA 敲低NSD2 或者通过同源重组的方式敲除易位的NSD2 基因都会抑制MM 的细胞增殖,促使细胞凋亡,抑制其肿瘤生成[13,18-19],而在无t(4;14)的MM 细胞系中过表达NSD2 后再进行荷瘤实验可以促进其成瘤性[13,18]。此外,在前列腺癌和子宫内膜癌中都发现NSD2 会提高转移相关基因TWIST1 基因体区域的H3K36me2 水平,促进基因转录,从而促进肿瘤转移和侵袭[20],而在t(4;14)的MM 中也存在NSD2 调控的TWIST1 的表达上调。
2.2 NSD2 与其它组蛋白修饰酶的相互作用 研究发现t(4;14)MM 细胞系中NSD2 过表达引起全基因组H3K36me2 水平升高的同时会引起H3K27me3 水平的降低,H3K27me3 是基因沉默的一个标记,虽然全基因组H3K27me3 水平的降低导致了多种基因的上调,但是特定位点的H3K27me3 水平又会升高,导致转录抑制,因此t(4,14)MM 细胞系对EZH2 抑制剂的处理敏感,EZH2 抑制剂可使t(4,14)MM 细胞系生长受到抑制[21-22]。Relja Popovic 研究组2004 年发表的工作发现在NSD2 表达水平低的MM 细胞系中EZH2结合水平低以及H3K27me3 比较少的位点,会在NSD2表达水平高的细胞系中出现EZH2 结合水平升高,许多在NSD2 表达水平高的细胞系中特有的EZH2 的基因组结合峰位于CTCF 位点,CTCF 位点是已知的绝缘子,NSD2 上调会在基因组上建立组蛋白修饰的边界,导致绝缘子另一侧基因EZH2 结合水平和H3K27me3水平的升高,表达受到抑制[22]。NSD2 的表达同时受到EZH2 的 调 控,miR-203,miR-26a 和miR-31 都是抑癌性microRNA,可以降低NSD2 的mRNA 的稳定性从而下调原癌基因NSD2 的表达,但EZH2 介导的H3K27me3 会抑制这类microRNA 的表达从而上调NSD2 的表达水平[23],这也说明了NSD2 和EZH2 之间的紧密联系。此外,NSD2 还可以与KAP1 复合物以及组蛋白去乙酰化酶结合发挥转录抑制作用[24-25]。
2.3 NSD2 与microRNA 的相互作用 MicroRNA 主要通过调控mRNA 的稳定性、影响靶基因的表达来发挥其生物效应,而microRNA 本身的表达又会受到表观修饰和转录因子的调控。D-J Min 等研究者通过转录组分析鉴定到miR-126*是NSD2 的一个靶标,而miR-126*又可以靶向c-myc mRNA 的3’非翻译区抑制其翻译过程,染色质免疫共沉淀实验发现NSD2 会和KAP1 以及HDAC1 共同结合在miR-126*的宿主基因启动子区域,形成H3K9me3 标记抑制其表达,因此,外源过表达miR-126*可以显著降低t(4;14)MM 细胞系的增殖速率[25]。此外,对正常浆细胞、MGUS 浆细胞以及MM 浆细胞的转录组分析鉴定到多种在MM 中异常转录的microRNA,如发挥抑癌作用的miR-196b、miR-135b、miR-320、miR-20a、miR-19b、miR-19a 和miR-15a 等的下调,发挥促癌作用的miR-17-92 簇、miR-221/222 簇等的上调[26],但这些micro-RNA 的异常转录是否受到NSD2 的调控有待进一步验证。
2.4 NSD2 与其他蛋白的相互作用 组蛋白修饰酶介导组蛋白的各种转录后修饰,可以影响染色质结构进而调节基因表达,但组蛋白修饰酶被靶向至特定基因位点或特定的基因组位置的机制尚不清楚,NSD2 亦然。因此,研究与NSD2 存在相互作用的蛋白对深入了解其功能和作用机制具有重要作用。蛋白免疫共沉淀和质谱结果显示NSD2 与IQGAP1、TIAM1 蛋白存在相互作用[27],这两个蛋白可以和β-catenin 相互作用参与到WNT 信号通路,基因表达谱显示NSD2 的敲低会显著下调CCND1 的表达,且染色质免疫共沉淀显示NSD2 会结合到Ccnd1 启动子区域[28],而CCND1又是β-catenin/Tcf4 复合物的下游靶基因,说明NSD2和β-catenin 之间可能存在相互作用共同调节其下游靶基因,而WNT/β-catenin 信号通路又在个体发育和前列腺癌中发挥重要作用,这与NSD2 异常会影响个体发育以及在前列腺癌中存在NSD2 的过表达相一致,说明了二者之间的联系。Yang P 等研究者发现在对阉割治疗有耐受的前列腺癌细胞中,NSD2 可以和NF-κB 直接相互作用共同激活其下游靶基因的表达,如IL-6、IL-8、VEGFA、BCL-2、BIRC5、c-MYC 和CCND1 等,染色质免疫共沉淀实验也证明NSD2 和NF-κB 在这些基因位点存在共定位,NSD2 的表达又会通过NF-κB 受到细胞因子如TNF-α、IL-6 的调控,NSD2 的过表达又会反过来与NF-κB 共转录激活其下游靶标,因此导致NF-κB 通路的持续激活[28],但这一蛋白相互作用以及调控环路是否在MM 中也存在有待进一步验证。最近的一项研究发现在t(4;14)MM 细胞系KMS11 中存在NSD2 和PARP1 的相互作用,后者可以催化NSD2 发生多聚ADP 核糖基化而抑制NSD2 在染色质上的结合[29]。
3 展望
现有研究表明,NSD2 是t(4;14)型MM 发生发展的核心调控因子,研究清楚NSD2 在t(4;14)型MM 中的具体致病机制将有助于治疗MM 的新药开发。研究表明在MM 中,存在NSD2 和EZH2 的相互作用,且利用小分子抑制剂对EZH2 进行干扰确实能抑制MM 细胞的增殖,这一作用能否被发展为临床治疗有待进一步研究,一些microRNA 的过表达也可以达到抑制MM 细胞增殖的效果,但microRNA 如何给药以及如何靶向特定类型细胞仍是个问题。此外,DNA 甲基化也是一个很重要的表观调控机制,在NSD2 介导的全基因组H3K36me2 的变化以及基因表达谱的变化中,是否存在和DNA 甲基化的相互作用值得研究,但目前已有研究表明利用5-aza 抑制DNA 的甲基化也能起到抑制MM 细胞增殖的作用[30],具体作用机制有待清楚阐明;最后,针对NSD2 的靶向抑制看似是最为直接的方式,所以需要进一步研究清楚NSD2 发挥其催化功能的具体方式和关键结构域,从而可以针对不同结构域来设计或筛选小分子抑制剂,近期的几项研究通过高通量筛选已经鉴定到多个靶向NSD2 的小分子抑制剂,能否被应用于临床治疗未来还需要进一步的研究。
猜你喜欢
热带作物学报(2022年7期)2022-08-06
中国农业科学(2022年14期)2022-07-26
保健与生活(2022年11期)2022-06-09
畜牧兽医学报(2022年3期)2022-03-30
中国畜牧兽医(2022年1期)2022-02-15
科学与生活(2021年16期)2021-11-25
保健与生活(2021年5期)2021-04-12
世界最新医学信息文摘(2020年19期)2020-03-31
安徽农业科学(2015年3期)2015-10-21
医学研究杂志(2015年9期)2015-07-01
