
裂殖壶菌诱变株高产DHA机理的转录组学分析
2020-01-16 02:12:44胡爱云龚定芳
中国油脂 2019年12期
胡爱云 ,龚定芳 ,李 莎
(1.无锡太湖学院 江苏省物联网应用技术重点建设实验室,江苏 无锡 214064;2.江南大学 生物工程学院,工业生物技术教育部重点实验室,江苏 无锡 214122)
随着科技的发展,人们对DHA的生理功能有了较为详细的了解[1-3]。DHA的来源主要分为两类:一类是传统鱼油DHA[4],主要从DHA含量较高的深海鱼类中提取获得;另一类是微生物发酵法获取DHA[5]。与其他海洋真菌相比,裂殖壶菌不仅能够大量积累油脂,还可以积累许多对人体有益的活性物质,具有生长速度快、DHA含量高等优势,市场前景广阔。裂殖壶菌藻油的安全性已经得到了美国FDA的认可,在我国卫生部2012年颁布的相关政策中提到允许在奶粉中添加裂殖壶菌藻油DHA[6]。在前期研究中,利用ARTP诱变技术获取一株高产DHA的裂殖壶菌SR21突变菌株,被命名为NA2[7],但其高产DHA的分子机理尚不明确。
转录组学已经被广泛用于各项科学研究,目前已经在裂殖壶菌的研究中得到运用。王康[8]使用基于RNA-seq的转录组学技术,对添加了萘氧乙酸或茉莉酸培养24 h的裂殖壶菌及正常培养24 h的裂殖壶菌进行转录组分析,根据基因差异表达的情况,证实了磷酸戊糖途径和糖酵解途径增强,氨基酸合成代谢途径减弱为油脂合成提供了更多的前体物质乙酰辅酶A,分析了提高DHA产量的机理。陈伟[9]基于比较转录组学,对甘油和葡萄糖两种碳源条件下的差异基因的表达进行了全面的分析,对甘油促进DHA大量积累的机理进行了解析。Ma等[10]基于转录组分析考察了低温情况下基因差异表达情况,结果表明低温导致脂肪酸合酶和3种多不饱和脂肪酸合酶亚基的表达分别下调约3.7倍和2倍。这些差异基因介导了冷适应的关键步骤,如信号转导、细胞成分生物发生、碳水化合物和脂质代谢等。
本研究将利用RNA-seq技术分析ARTP诱变菌株NA2高产DHA的分子机理,并利用荧光定量PCR对基因的差异表达情况进行验证。
1 材料与方法
1.1 实验材料
菌种:裂殖壶菌菌株(SchizochytriumlimacinumSR21),购自美国菌种保藏中心(ATCC),-80℃保存于ATCC By+790培养基中。活化培养基:ATCC By+790固体培养基。种子培养基:葡萄糖 30 g/L,酵母粉6 g/L,鱼粉蛋白胨6 g/L。摇瓶培养基:参考文献[11] 。诱变菌株NA2,在前期研究中由ARTP诱变得到[7]。
1.2 转录组分析方法
1.2.1 RNA测序及数据处理
RNA的提取:取摇瓶培养96 h的原始菌株SR21和诱变菌株NA2发酵液4 mL,10 000 r/min离心3 min,利用UNIQ-10柱式Trizol试剂盒提取总RNA。
RNA-seq步骤参照文献[12]。
测序数据的处理:Illumina HiSeq上机测序得到原始测序序列(Raw Reads),为了确保信息分析准确性,必须对原始测序序列过滤,首先检查测序错误率分布及A/T/G/C 含量分布,然后去除带接头的、质量低的以及N(N表示无法确定碱基信息)所占比例大于10%的测序序列,得到过滤后序列。再采用Trinity[13]对过滤后序列进行拼接获得转录本,作为后续分析的参考序列,用Corset[14]程序对转录本进行层次聚类,以聚类后得到的每条基因中最长的转录本作为该基因的代表,称为Gene,以此进行后续的分析。
1.2.2 测序数据分析
参照七大数据库(参照文献[15] )对测序的结果进行基因功能注释。
采用DESeq2[16]进行差异基因筛选,筛选阈值为差异显著性p值小于0.05(padj<0.05)且对差异倍数取以2为底的对数之后的值大于1(|log2FoldChange|>1)。利用Pathway显著性富集[17]进一步对差异表达基因进行Kegg富集分析。
1.2.3 实时荧光定量PCR验证(RT-qPCR)
按照1.2.1所述提取总RNA,再将其反转录成cDNA,反转录试剂盒为南京诺唯赞公司的HiScript III RT SuperMix for qPCR(+gDNA wiper)。对cDNA进行梯度稀释,将稀释后的模板进行RT-qPCR扩增,验证引物的扩增效率。采用染料法进行RT-qPCR,验证差异基因的差异表达倍数。其中以裂殖壶菌18S rRNA为内参,引物设计采用线上设计,网址为https://sg.idtdna.com/scitools/Applications/RealTimePCR/。RT-qPCR反应体系如表1所示。检测仪器为CFX96 Real-Time system,设置PCR的程序为:95℃预热30 s,95℃变性5 s后60℃退火延伸30 s并循环40次。融解曲线参数参照试剂盒设置。

表1 RT-qPCR反应体系
最终以2-ΔΔCt来表征基因的差异表达倍数,其中Ct值是指在PCR过程中,扩增产物的荧光信号达到设定的阈值时所需要的循环次数[18]。每个样品重复3次。
2 结果与讨论
2.1 总RNA样品质量评估
用摇瓶将原始菌株SR21和诱变菌株NA2培养96 h,分别取发酵液4 mL,提取总RNA。利用以下方法检测所获取的总RNA的质量:①琼脂糖凝胶电泳检测RNA降解程度、污染程度;②Nanodrop检测RNA的纯度(OD260/OD280);③Qubit对RNA浓度进行准确定量;④利用Agilent 2100精确检测RNA的完整性。进行两次平行实验,具体结果见表2。
通常认为,OD260/OD280处于1.8~2.2之间,OD260/OD230大于2时核酸纯度较高,RNA完整性(RNA Integrity Number,RIN)越接近10则表示RNA越完整,28S/18S越接近2则表示RNA质量越好[19]。由表2可知,综合各项考察指标,本研究中所获取的4个RNA样品均满足RNA-seq的要求。
2.2 测序数据质量评估
Illumina HiSeq上机测序得到原始测序序列,检查碱基错误率分布及A/T/G/C 含量分布。过滤原始数据,去除带接头的、低质量的以及N(N表示碱基信息没办法确定的)所占比例大于10%的序列,最终得到过滤后数据。具体的评估计算结果如表3所示。
注:Q20、Q30表示碱基质量大于20或30所占的比例;GC表示的是检测片段中G和C的含量。
从表3可以看出,碱基错误率低于0.1%,过滤后序列数据量大,Q20和Q30较大,均高于90%。
进一步对过滤后序列进行转录本(Transcript)拼接和Corset层次聚类,得到基因序列(Unigene),最终得到的序列信息以FASTA格式储存。拼接转录本与基因序列长度分布如图1所示。
由图1可知,Unigene共包含了19 213个基因片段,长度在2 000 bp以上的共有5 191个。
2.3 基因功能注释
为得到更加全面的基因功能信息,本研究参照七大数据库(Nr、Nt、Pfam、Go、Kog、Swiss-prot、Kegg)对最长的转录本进行了基因功能注释。选择与研究目的相近、注释成功率高的5个数据库绘制了维恩图,见图2。从图2可以看出,能够在5个数据库中被正确注释的基因共有1 042个,同时仍有大量未能被正确注释的基因存在。注释率较低的原因有以下两点:①转录组数据里面有一部分是非编码序列;②裂殖壶菌是研究较少的菌种,现有数据库中该菌种及其相关菌种注释较少。

图2 基因功能注释结果维恩图
(1)Nr注释:通过与Nr库进行比对注释,既能够清楚地知道研究物种的基因序列与亲缘关系较近的物种基因序列的相似性,也可以得到研究物种基因的功能信息。对比结果为:裂殖壶菌菌株与球孢白僵菌(Beauveriabassiana)、马尾松毛虫菌(Spizellomycespunctatus)、裂孢蛙粪霉菌(Basidiobolusmeristosporus)、根内球囊霉(Rhizophagusirregularis)、节水霉菌(Gonapodyaprolifera)以及其他菌种的基因序列相似性分别为14.7%、 5.5%、 5.4%、 3.2%、 3.1%及68.1%。
(2)Go是一套国际标准化的用于描述基因功能的分类系统。Go分为3个大类:生物过程(Biological Process,BP)、细胞组成(Cellular Component, CC )、分子功能(Molecular Function,MF)。Go注释对基因编码的产物进行深度解析,分别探究其参与的生物过程、具有的分子功能和所处的细胞环境。对基因进行Go注释以后,将注释成功的基因依照下一层级进行分类,结果如图3所示。从图3可知:归类到生物过程的这类基因中,大量的基因参与细胞内过程类别、代谢过程和单组织过程类别;参与细胞组成的基因主要集中在细胞组成、膜组成和细胞器组成3个部分;分子功能类别中,具有结合和催化活性功能的基因较多,远高于其他功能下的基因数目。
(3)Kegg数据库:该数据库是一个系统性分析基因产物功能以及其参与细胞中哪些代谢途径的数据库。对基因进行Kocc(Kegg ortholog)注释后,可以根据其参与的Kegg代谢通路分为以下几个类别:细胞过程(A,Cellular Processes)、环境信息处理(B,Environmental Information Processing)、遗传信息处理(C,Genetic Information Processing)、代谢(D,Metabolism)、有机系统(E,Organismal Systems),结果见图4。从图4可知,参与代谢过程的基因所占的比例比较高,主要集中在碳水化合物和氨基酸的代谢过程中。
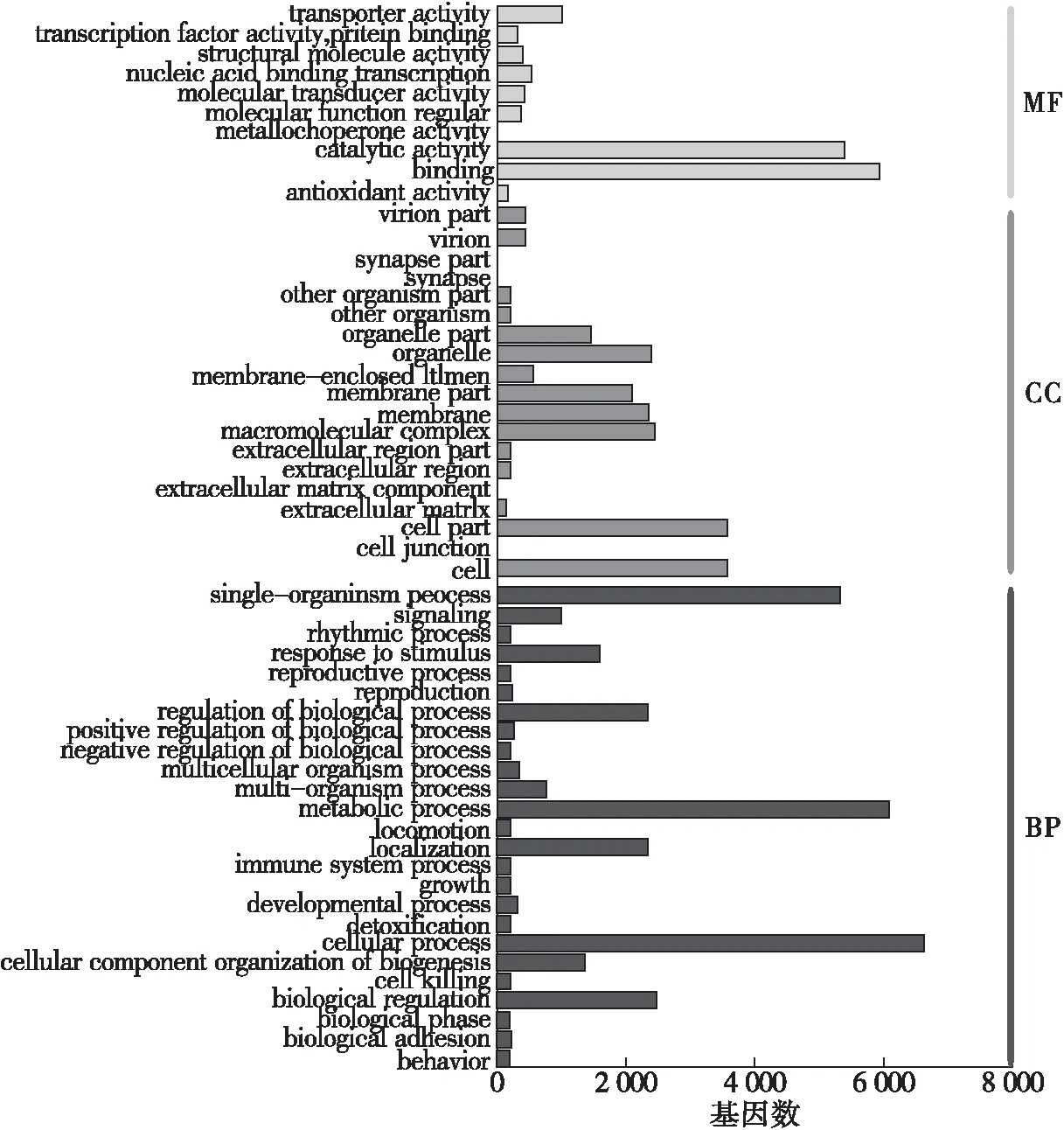
图3 Go分类图
2.4 差异基因表达
2.4.1 基因差异表达分析
根据每个样品基因表达水平的结果,用DESeq2[16]进行分析,检测样品之间的差异表达基因(Differentially Expressed Gene,DEG),制作火山图(略),差异基因筛选阈值为padj<0.05且|log2FoldChange|>1。对于差异基因,如果基因的log2FoldChange>0,则认为该差异基因上调,反之,认为该差异基因下调。以-log10(padj)的数值大小来表征差异的显著性。与对照组(原始菌株SR21)相比,实验组(诱变菌株NA2)共有314个基因上调,3个基因下调,且上调部分基因上调的幅度极大。
绘制基因表达的维恩图,见图5。
图5直观地展现了2个实验样品之间独有和共有的表达基因数目。两组样品共有14 278个共同表达基因,实验组有4 025个特有表达基因,对照组有889个特有表达基因。

图4 Kegg分类图

注:以FPLM>0.3为基因表达的标准,每百万碎片中来自某一基因每千碱基长度的碎片数目。
图5 基因表达维恩图
2.4.2 Kegg富集分析
在生物体中,许多基因之间相互协调,共同发挥作用,行使某项生理功能, 通过Pathway显著性富集能够明确这些差异表达基因具体参与的是何种生化代谢途径和信号转导途径。富集结果如表4所示。
由表4可知,共有19个上调表达的基因被富集到17个代谢途径中,主要包括氨基酸代谢、能量代谢等。同时可以看出,同一个基因可能参与几个代谢途径, 最终得到9个差异基因,分别是ECHS1、TPIA、SETD1、CHK2、SIR2、MSI、REA1、UBE2Z、PURL。
ECHS1(烯脂酰辅酶A水合酶短链1)是脂肪酸分解代谢β氧化通路的关键酶[20],同时参与支链氨基酸氧化。从表4可知,ECHS1同时参与赖氨酸、β-谷氨酸、色氨酸、缬氨酸、亮氨酸和异亮氨酸等多种氨基酸的降解。氨基酸的降解可生成更多的乙酰-CoA,为脂质合成提供更多的前体物质。另外ECHS1还参与脂肪酸的延长和降解途径,β氧化是一个脂肪酸改造的过程,根据机体自身的需要,一些长链脂肪酸会分解,释放能量,然后合成长度适宜的脂肪酸。相比于饱和脂肪酸的β氧化,DHA的β氧化还需要异构酶和还原酶的参与[21]。ECHS1基因上调说明细胞内脂肪酸代谢活跃,脂肪酸合成速度太快,前体物质无法满足,因此一些脂肪酸被分解以提供更多的能量和原材料供某些特殊脂肪酸的合成[22],这可能是诱变菌株NA2高产DHA的原因。
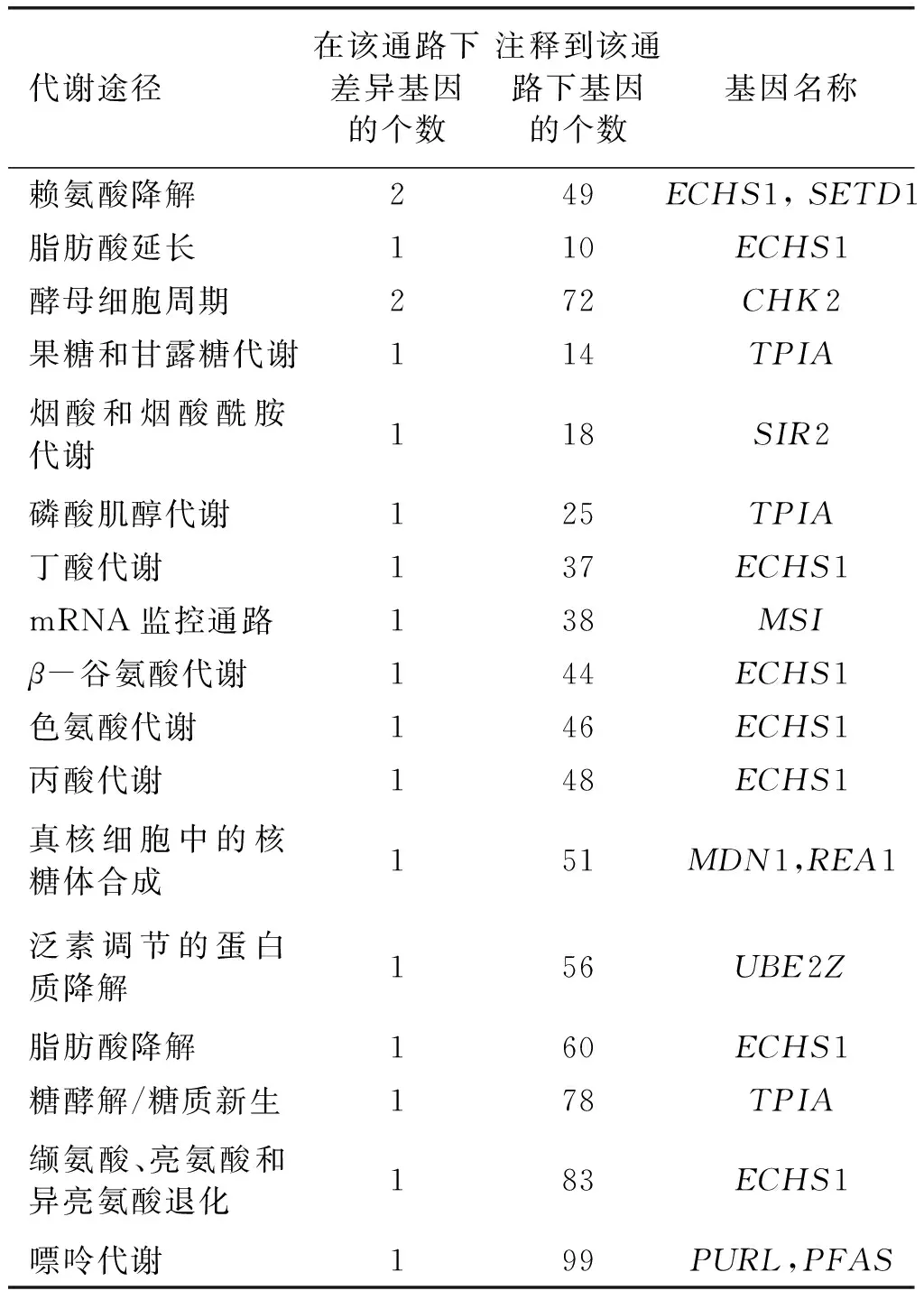
表4 差异基因Kegg显著性富集列表
TPIA(磷酸丙糖异构酶)是糖酵解途径中的关键酶之一,在糖酵解和脂肪酸合成等途径中具有非常重要的作用。TPIA催化磷酸二羟丙酮(DHAP)发生异构化形成3-磷酸甘油醛(G-3P)并用于后续代谢过程,在这个过程中NAD+转化为NADH,为脂质合成提供能量。
SETD1是组蛋白赖氨酸甲基转移酶SET1家族的成员,主要对赖氨酸和精氨酸的残基进行组蛋白甲基化修饰。另外SETD1和CHK2都与DNA的修复有关。细胞周期检测点CHK2基因上调可加强对G2/M期的检查,防止带有基因组 DNA 损伤的细胞进入有丝分裂,维持真核细胞基因组的稳定性。
SIR2是一组进化上高度保守的NAD+依赖的组蛋白去乙酰化酶[23],控制细胞寿命、转录调控、能量代谢、抗氧化应激。研究表明该酶参与染色质沉默,染色质沉默对维持染色体结构稳定和基因调控起重要作用[24]。MSI是一种RNA结合蛋白,参与细胞不对称分裂及基因转录调控,抑制细胞凋亡和分化。REA1 是核糖体合成因子,与MSI共同作用实现了基因的高效表达。
UBE2Z:泛素介导的蛋白质水解过程,特异性强,效率高,需要消耗能量。待降解的蛋白质首先被泛素标记,然后进入细胞的蛋白酶复合的活性结合位点,进而被降解成短肽。PURL:调控嘌呤代谢途径关键酶基因表达水平,影响经AMP→ADP→ATP代谢途径,促进能量的产生。UBE2Z和PURL基因上调,说明细胞内代谢活跃,表明细胞增殖迅速,这可能与生物量的提高有关。
2.5 RT-qPCR验证
转录组测序分析的结果往往需要通过RT-qPCR进一步验证,从转录组测序结果中筛选出9个基因进行RT-qPCR验证,以裂殖壶菌18S rRNA为内参,设计引物如表5所示。

表5 引物设计序列
续表5

基因名称引物序列(5'-3')SIR2F:GATAA CGGACCTAC TGCCATGR:CTTTCCTCAGCACGATCTACGMSIF:ACCTCCCTACAATCAGTCTCGR:TCCAAATTCCAGAGCTGTCAGREA1F:TGGTAATCGGTATCTGTGAGT-TGR:ACCAGTCGTTTCATTGTCCTCUBE2ZF:TCGCCAAGTACCAAGTAAAGAGR:CCCTGAGTATCCCAATTGTCCPURLF:TGGTAATCGGTATCTGTGAGT-TGR:ACCAGTCGTTTCATTGTCCTC
将RT-qPCR的结果整理后得到基因相对表达差异倍数如图6所示。

图6 RT-qPCR基因相对表达差异倍数
为了验证转录组测序结果的准确性,以RT-qPCR中-ΔΔCt值与转录组分析的结果log2ratio对比,结果如图7所示。由图7可知,差异基因的表达量倍数上有所差异,但是在变化趋势上是一致的。说明转录组测序的结果是可信的。
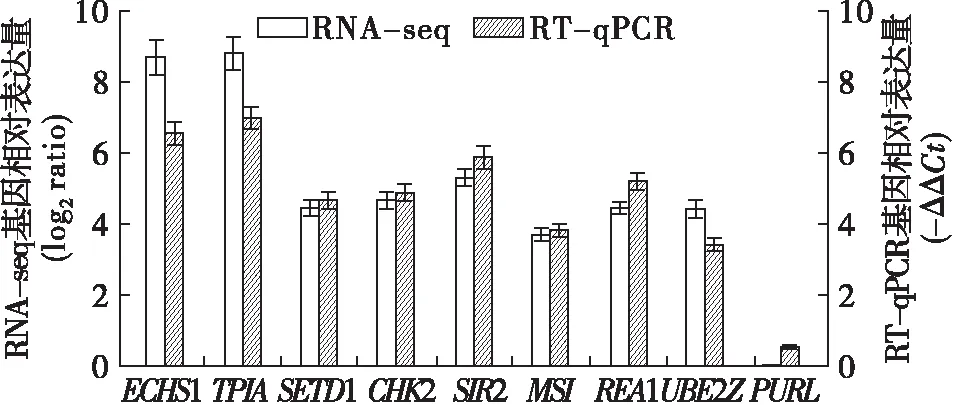
图7 相关酶基因RNA-seq和RT-qPCR基因相对表达量对比
RT-qPCR的结果进一步证明了脂肪酸代谢、氨基酸代谢途径中一些关键基因的突变,引起转录水平的变化。维持基因组稳定性和实现高水平转录等的相关基因上调共同促进了DHA的积累。以上结果表明ARTP这种新型的诱变方式是简单有效的,产生了较多的突变位点,同时利用丙二酸平板成功筛选获得高产菌株,在维持葡萄糖浓度恒定,间歇补加氮源的补料分批发酵策略下提高了DHA的产量。
3 结 论
采用转录组分析的方法揭示了诱变菌NA2中DHA产量提高的分子机理:与原始菌株相比,诱变菌NA2共有314个基因上调,3个基因下调;上调的基因主要参与氨基酸代谢和能量代谢,为多不饱和脂肪酸的积累提供更多的乙酰-CoA和NADPH。
猜你喜欢
动物医学进展(2024年4期)2024-04-10 01:50:04
中国生殖健康(2020年5期)2021-01-18 03:00:06
心电与循环(2020年1期)2020-02-27 07:48:24
发明与创新·大科技(2019年5期)2019-07-31 07:35:28
中国生殖健康(2018年5期)2018-11-06 07:15:56
科学之谜(2017年8期)2017-09-14 21:46:33
飞碟探索(2017年2期)2017-02-13 21:54:30
中国洗涤用品工业(2015年8期)2015-02-28 19:02:49
湖北农业科学(2014年3期)2014-07-21 10:25:00
食品科学(2013年19期)2013-03-11 18:27:44
