
Evolution of specific RNA aptamers via SELEX targeting recombinant human CD36 protein: A candidate therapeutic target in severe malaria
2020-01-10 06:31NikAbdulAzizNikKamarudinJudyNurAishaSatNurFatihahMohdZaidiKhairulMohdFadzliMustaffa
Nik Abdul Aziz Nik Kamarudin, Judy Nur Aisha Sat, Nur Fatihah Mohd Zaidi, Khairul Mohd Fadzli Mustaffa
Institute for Research in Molecular Medicine (INFORMM), Health Campus, Universiti Sains Malaysia, 16150, Kubang Kerian, Kelantan, Malaysia
ABSTRACT Objective: To isolate and characterize RNA aptamers that are specific to human CD36 protein using systematic evolution of ligands by exponential enrichment (SELEX) technology to identify candidates for adjunct therapy to reverse the binding of Plasmodiuminfected erythrocytes. Methods: RNA aptamers were isolated using nitrocellulose membrane-based SELEX and binding analysis was screened using an electrophoretic mobility shift assay and enzyme-linked oligonucleotide assay. Results: Thirteen cycles of nitrocellulose membrane-based SELEX yielded three aptamers (RC60, RC25, RC04) exhibiting high binding against CD36 protein as shown on electrophoretic mobility shift assay. The sequence analysis revealed a G-quadruplex sequence within all the isolated aptamers that might contribute to aptamer binding and thermodynamic stability. The specificity assay further showed that RC60 and RC25 were highly specific to CD36. The competitive inhibition assay demonstrated that RC60 and RC25 shared a similar binding epitope recognized by mAb FA6-152, a specific monoclonal antibody against CD36. Conclusions: RC60 and RC25 are promising candidates as anticytoadherence for severe malaria adjunct therapy.
KEYWORDS: RNA aptamers; SELEX; CD36 protein; Severe malaria; Adjunct therapy; Cytoadherence
1. Introduction
Malaria is one of the significant parasitic infection in human causing 429 000 deaths worldwide, mostly in the endemic areas especially in Africa region, followed by the South-East Asia region and the Eastern Mediterranean region[1]. Most of the cases are affecting children under five years old. Even though the malaria mortality rate among children is reduced to 35% from 2010 until 2015, but the disease remains a significant killer, claiming the life of 1 child every 2 minutes[2]. Plasmodium falciparum is the most significant agent of malaria in human and responsible for the enormous burden of global mortality and morbidity[3]. A unique characteristic of the mature infected erythrocytes (IEs) to sequestrate the microvascular surface protein has been associated with the development of severe malaria (SM), which leads microcirculatory obstruction, impaired tissue perfusion and inflammatory cells activation[4]. This sequestration mechanism allows the parasite to escape from spleen-dependent killing mechanisms and act as a determinant for adhesion-based complications of malaria infections such as cerebral malaria and pregnancy-associated malaria[5].
The CD36 protein is a multiligand scavenger receptor that facilitates the binding and uptake of a wide variety of particulate ligands such as oxidized low-density lipoproteins, bacteria, β-amyloid plaque, and apoptotic cells by macrophages[6]. The CD36 protein is involved in many biological processes including mediating the sequestration of IEs in microvascular capillaries, thrombosis, anti-angiogenesis, inflammation, phagocytosis and endocytosis[7,8]. The CD36-mediated cytoadherence of IEs conceivably will contribute to the dysfunction of vital organs such as kidney, lung, and liver by impairing the microcirculatory blood flow in malaria patients[8]. Interestingly, this protein is expressed at a low level in the brain and uninducible by inflammatory cytokines such as tumor necrosis factor alpha (TNF-α). The binding of IEs to CD36 receptor protein commonly caused uncomplicated malaria but it also can indirectly support the cytoadhesion of the IEs to the microvascular brain which can lead to the development of SM through binding to multiple receptors including endothelial protein C receptor[9]. The expressed CD36 on the platelet molecules has been reported to facilitate the platelet clumping and endothelial microvascular obstruction due to sequestered IEs which strongly associated with SM[4]. A recent report using in vivo study of Plasmodium berghei infection in mice showed that sequestration of the parasite is the CD36-dependent type that can promote parasite growth and survival in the host where sequestration occurred at the CIDRα2-6 domain of PfEMP1[10,11]. Previously, monoclonal antibodies (mAb) FA6-152 and OKM5 have been reported to bind to a specific immune-dominant of 153-183 and 139-184 amino acid of the CD36 protein, respectively to inhibit the cytoadherence of IEs[12]. Even though the successful finding of mAb to inhibit and reverse the cytoadherence, there are some limitations of the use of mAb in immunotherapy including high immunogenicity, expensive cost, batch to batch variation, larger size, thermal instability and chemical modification[13].
In past 20 years, Ellington and Szostak had discovered new small molecules that can be used for therapy and diagnostic which mimic mAb called aptamer[13]. The aptamer is a synthetic oligonucleotide in the form of single-stranded DNA (ssDNA) or RNA, which could bind to a variety of targets at high specificity and affinity. Aptamers bind to their target via an “induced fit” mechanism, similar to antibodies[14]. Even though both DNA and RNA aptamer can form complex structures, the RNA aptamer can form a more diverse tertiary structure as RNA contains 2’-OH group on their ribose sugar that contributes to tighter and specific binding to the target[15]. The modification of RNA aptamer at 2’OH with 2’ fluoro, 2’amino, and 2’O-methoxy has been found to greatly increase resistance to nuclease environment within the bloodstream. Systematic evolution of ligands by exponential enrichment (SELEX) technology is a method used to isolate aptamers, with high affinity and specificity to the target, and having a dissociation constant in the low nanomolar to picomolar level[14,15]. Aptamers have more advantages than antibodies regarding production, stability, binding affinity, small size, and less immunogenicity[16]. Previous aptamer development for malaria treatment is focused on disruption of rosette formation and heme metabolism[13]. Unfortunately, an aptamer targeting the host endothelial surface receptors to block or reverse the binding of IEs has not yet been explored.
The present work describes the use of a SELEX strategy to isolate RNA aptamers against the recombinant (rhCD36). The isolated potential RNA aptamer candidates could be further studied on their potential to inhibit and reverse the IEs binding to human endothelial surface CD36 protein.
2. Materials and methods
2.1. Preparation of RNA library
The aptamer library (5’-GCGAATTCTGGGGCGATATATCC(N40)CCAAATAGCCGAATTCGCACG-3’), forward primer (5’-GCTA ATACGACTCACTATAGGCGAATTCTGGGGCGATATATCC-3’) and reverse primer (5’-CGTGCGAATTCGGCTATTTGG-3’) were synthesized by the Integrated DNA Technologies (United Kingdom) and PAGE purified. The T7 promoter region (underlined) and N40 denotes 40 consecutive A, G, C or T nucleotides at 1:1:1:1 ratio, thus providing for approximately 1015random RNA library. Amplification of ssDNA aptamer library was done by mixed 1×Green GoTaq®Flexi Buffer, 1.5 mM MgCl2, 0.2 mM dNTPs, 0.5 µM forward primer, 0.5 µM reverse primer, 10 nM oligonucleotide and 5 units GoTaq®DNA Polymerase (Promega, United States) in 100 µL total reaction. Then, the reaction mixture was amplified using the following condition; predenaturation at 94 ℃ for 5 min, 5 cycles of denaturation at 94 ℃ for 30 s, annealing at 61 ℃ for 30 s, elongation at 72 ℃ for 30 s and postelongation at 72 ℃ for 7 min using Veriti®Therma Cycler (Thermo Fisher Scientific, US). The PCR product was ethanol precipitated and followed by transcription using AmpliScribe™T7-Flash™Transcription Kit (Epicentre Biotechnology, USA) according to the manufacturer’s protocol. Briefly, the transcription reaction was performed in 20 µL containing 1× of AmpliScribe™T7-Flash™Reaction Buffer, 9 mM of each ATP, CTP, GTP, and UTP, 20 units of RiboGuard™RNase Inhibitor (40 U/µL), 1 µg of dsDNA sample and 20 units of AmpliScribe™T7-Flash™Enzyme Solution (10 U/µL). The reaction mixture was mixed and incubated at 37 ℃ for 16 h (overnight) using AccuBlock Digital Dry Bath (Labnet International. Inc, Edison). On the following day, the reaction tube was added with 10 units of RNase-free DNase I and incubated again at 37 ℃ for 20 min to remove the dsDNA that existed in the reaction mixture. The reaction mixture was purified by using phenol: chloroform (24:1) and precipitated by using ethanol.
2.2. Selection of RNA aptamer
SELEX experiment was performed using nitrocellulose membrane filter immobilization methods as described by Hall and colleagues[17]. Briefly, the total RNA pool was resuspended in 100 µL of 1×SELEX buffer (SB) (137 mM of NaCl, 2.7 mM of KCl, 10 mM of Na2HPO4and 1.7 mM of KH2PO4, pH 7.4) and heated at 95 ℃ for 2 min, followed by cold down at room temperature for 10 min to allow RNA refolded into the secondary structure. The RNA pool was subjected for negative selection by filtering through pre-equilibrated (in 1×SB) 0.45 µm nitrocellulose membrane filter (Millipore, USA) using ‘pop-top’ filter holder (Whatman, United Kingdom) to remove RNA/filter bound and collect the filtrate (non-filter binder). The negative selection was performed in every SELEX cycle. During first SELEX cycle, 700 nM of the filtrate was mixed 20 nM of yeast tRNA (Life Technologies, USA) and 650 nM rhCD36 protein (R&D system, United Kingdom) in 100 µL of 1×SB. Then, the reaction was incubated at room temperature for 30 min on MACSmix tube rotator (MiltenyiBiotec, Germany). The binding reaction was filtered through a pre-wet 0.45 µm nitrocellulose membrane filter. After washed with 2× volume of 1×SB, the nitrocellulose membrane filter was collected and transferred to a new 1.5 mL tube. The RNAs/protein complex that bound to nitrocellulose membrane filter was recovered by heating in 400 µL elution buffer containing 7 M Urea at 95 ℃ for 5 min. The bound RNA was transferred into a new centrifuge tube followed by ethanol precipitation. The stringency of selection was done by reducing the protein concentration and the incubation time and increasing the yeast tRNA concentration. Addition of nucleic acid competitors such as yeast tRNA or spermidine has the advantage to reduce nonspecific binding of RNA library[18]. The SELEX experiments were performed until significant binding affinity towards the target molecule was observed. In this study, the amount of the nucleic acid retained as protein/RNA complexes on the surface of the nitrocellulose membrane filter were collected, reverse transcribed, amplified and gel electrophoresed before quantified using the ImageJ software[19].
2.3. Amplification of bound aptamer by RT-PCR
The recovered RNA was reverse-transcribed using reverse transcriptase (Promega, United State). The reaction mixture was conducted in 20 µL containing 1×AMV reverse transcriptase, 0.5 µM of reverse primer, 1.0 mM of dNTPs and 20 units of AMV reverse transcriptase. The reaction mixture was firstly heated at 95 ℃ for 2 min following cold down at room temperature for 10 min before dNTPs and enzyme were added. The reaction mixture was incubated at 42 ℃ for 1 h. The first cDNA was amplified using PCR. The PCR cycle was optimized until the band of the right size appeared. The PCR product was confirmed by electrophoresis of 10 µL of the PCR mixture on 2% (w/v) agarose gel and visualized using GeneFlash image analyzer (Syngene, USA). The PCR product was ethanol precipitated and directly used for the in vitro transcription using the AmpliScribe T7-Flash Transcription Kit. The isolated RNA was used for the next cycle of SELEX selection.
2.4. Cloning and sequencing
After 13 rounds of selection, the RT-PCR product was cloned using the TOPO TA Cloning Kit (Invitrogen, USA). There were 66 colonies that were randomly selected from positive individual colonies by using blue and white screening and plasmids were extracted using DNA-spin Plasmid DNA Extraction Kit (iNtRON Biotechnology, Korea). The plasmid was sent for sequencing using M13 forward primer (5’-GTAAAACGACGGCCAGT-3’) and M13 reverse primer (5’-CAGGAAACAGCTATGAC-3’) (First BASE Laboratories Sdn Bhd, Malaysia).
2.5. Prediction of secondary structure
The sequencing results were analyzed and aligned using ClustaW Multiple Alignment provided by MEGA 6 software (Version 6.06). The RNAs were clustered based on sequence alignment and phylogenetic analysis (Neighbor-Joining Tree) provided by MEGA 6 software. The online mfold software (http://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form) was used to predict the secondary structures of RNA aptamers. The secondary structure is essential to predict the binding region of the aptamer with the target and its functionality. Therefore, the analysis was conducted using free online mfold software[20]. The predicted secondary structures that have the lowest Gibbs free energy were selected because it will form the most stable secondary structure predicted by the software[20]. The aptamer sequences were further analyzed their motif consensus using online MEME (Multiple Em for Motif Elicitation) software at http://meme-suite.org/tools/meme (Version 4.12.0). The E-value generated by the software with the value less than or equal to 0.05 indicates that the motif identified is significant[21]. The online QGRS Mapper (http://bioinformatics.ramapo.edu/QGRS/analyze.php) was used to determine the G-quadruplex sequence of isolated RNA aptamer[22].
2.6. Electrophoretic mobility shift assay (EMSA)
The plasmids of the selected aptamer cluster were amplified by using specific primers as described above. The products were gel purified and used for in vitro transcription. Binding activity of selected aptamers and target protein-ligand was analyzed using fluorescence-based EMSA. This method uses two fluorescent dyes for detection, which are PeqGreen nucleic acid stain for RNA and SYPRO Ruby protein gel stain for protein. The binding assay was prepared by mixing 1 µM of selected RNA aptamer clusters (initially denatured at 95 ℃ for 2 min in 1×SB before cooling to room temperature for 10 min) with 0.5 µM of rhCD36. The binding reactions were then incubated at room temperature for 20 min. Following the addition of 4 µL of 6×loading dye, the RNA and protein complexes were separated from the free RNA using 5% (v/v) native PAGE. After electrophoresis at 140 V for 40 min in 0.5×Tris-Borate-EDTA (TBE) buffer, the gel was stained with a 1×PeqGreen nucleic acid stain in 0.5×TBE buffer for 10 min. After that, the gel was stained with SYPRO Ruby Protein stain for 3 h followed by destaining using destaining solution [10% (v/v) 2-Isopropanol, 10% (v/v) glacial acetic acid] for 1 h. The gel was visualized using a standard 300 nm UV transilluminator for both stains.
2.7. Semi-quantitative RT-PCR (sqRT-PCR) assay
The semi-quantitative RT-PCR assay was carried out to further evaluate the binding ability of isolated RNA aptamer to the target protein. This assay was used to quantify the bound RNA to the target by measuring the intensity of reverse transcribed and amplified product from the gel electrophoresis using ImageJ software[19]. The binding reactions were prepared at a fixed concentration of RNA and rhCD36 protein for all selected RNA aptamers. RNA was diluted to a final concentration of 1 µM and initially denatured at 95 ℃ for 2 min in 1×SB before cooling down to room temperature for 10 min. Subsequently, 0.5 µM of target proteins were added to corresponding tubes, and the reaction mixture was incubated at room temperature for 20 min. The RNA/protein complexes were separated using a nitrocellulose membrane filter. The eluted (bound) and filtrate (unbound) RNA/s were reverse transcribed and amplified. The amplification was performed at the 13th PCR cycle to avoid overamplification of PCR products. The PCR product was confirmed by electrophoresis of 10 µL of the PCR mixture on 2% (w/v) agarose gel and visualized using an image analyzer. The band’s intensity of amplified products was measured using ImageJ software (Version 1.46r).
2.8. Determination of dissociation constant value
The modified agarose-based EMSA assay was used to determine dissociation constant (Kd) as described by Ream et al[23]. Briefly, the series of rhCD36 protein concentrations were used from 0, 25, 50, 100, 250 and 500 nM and fixed concentration of RNA (100 nM). The complexes were resolved on 1% (w/v) agarose gel containing 1×Diamond Gel Stain (Promega, United States) (in 0.5×TBE buffer), ran 150 volts for 10 min. The gel was visualized using a standard 300 nm UV transilluminator. The signal intensity of bound and free RNA was measured using the ImageJ program and calculated Equation 1. The equilibrium dissociation (Kd) constant was calculated using nonlinear, curve-fitting algorithm [Y = Bmax*X / (Kd + X)] (GraphPad Prism 7.0 Software).
Equation 1: Percentage of fraction bound aptamer.
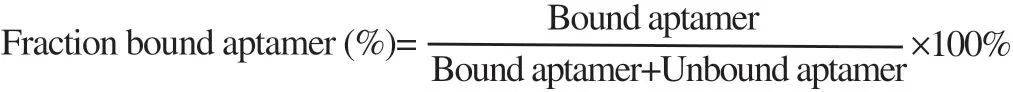
2.9. The specificity of isolated RNA aptamers
The RNA aptamer with a polyA tail was synthesized using a polydT reverse primer (5’-TTTTTTTTTTTTTTTTTTTTTTTTCG TGCGAATTCGGCTATTTGG-3’). Enzyme-linked oligonucleotide assays (ELONA) based binding assay was conducted in order to determine the binding specificity of rhCD36-isolated RNA aptamer cluster to bovine serum albumin (BSA) protein and ICAM-1 protein. Briefly, 1 µg/mL rhCD36, recombinant human intercellular adhesion molecule 1 (rhICAM-1) (R&D system, United Kingdom) and BSA (Sigma-Aldrich, United States) (diluted in bicarbonate/carbonate coating buffer, pH 9.6) were coated on 96 wells microplate at 37 ℃ for 1 h. The plate was then washed with ELISA washing buffer (1×PBS, pH 7.4 containing 0.02% (v/v) Tween-20) three times using ELISA plate washer. Afterward, the plate was blocked with 2% (w/v) non-fat milk (Oxoid, United Kingdom) diluted in ELISA washing buffer. After that, the mixture of 100 nM of polyA tail RNA aptamer and 100 nM of biotinylated polydT (5’-/BiosG/TTTTTTTTTTTTTTTTTTTT3’) (previously diluted in 1×SB denatured at 95 ℃ for 2 min and reannealed at room temperature for 10 min) was added and the plate was incubated at 37 ℃ for 1 h. The bound RNA was detected using HRP-conjugated streptavidin (Thermo Fisher Scientific, United States) (diluted 1:10 000 in 1×PBS, pH 7.4). After washing five times, the color was developed by adding TMB substrate and reaction was stopped using 2N HCl. The absorbance was read at 450 nm using SpectraMax M5 (Molecular Device, United States).
2.10. Competitive inhibition assay
Ten µg/mL of avidin (Thermo Fisher Scientific, United States) (diluted in bicarbonate/carbonate coating buffer, pH 7.4) was coated on 96 wells microplate at 4 ℃ overnight. The plate was then washed with ELISA washing buffer (1×PBS, pH 7.4 containing 0.02% (v/v) Tween-20) three times using ELISA plate washer. The plate was then blocked with 2% (w/v) non-fat milk diluted in ELISA washing buffer. Subsequently, the mixture of 100 nM of polyA tail RNA aptamer and 100 nM of biotinylated polydT (5’-/BiosG/TTTTTTTTTTTTTTTTTTTT3’) (previously diluted in 1×SB, denatured at 95 ℃ for 2 min and reannealed at room temperature for 10 min) was added and the plate was incubated at 37℃ for 1 h. After washing, the mixture of 1 µg/mL of rhCD36 and 1 µg/mL of mAb FA6-152 (Abcam, United Kingdom) was added and the plate was incubated at 37 ℃ for 1 h. As the control, the polyA tail RNA aptamer was allowed to bind with rhCD36 in the absence of mAb FA6-152. The bound rhCD36 was detected using HRP-conjugated anti-human IgG (Thermo Fisher Scientific, United States) (diluted 1:100 000 in 1×PBS, pH 7.4). After washing five times, the color was developed by adding TMB substrate and reaction was stopped using 2N HCl. The absorbance was read at 450 nm using SpectraMax M5 (Molecular Device, United States).
2.11 Statistical analysis
Data analysis was done using Two-way ANOVA and unpaired t-test using GraphPad Prism (Version 7.0). Error bar indicates standard deviation(±SD) of the triplicate experiments and P-value less than 0.05 was considered statistically significant.
3. Results
3.1. Selection of RNA aptamers against human rhCD36
Isolation of RNA aptamers specifically bind to recombinant human endothelial surface receptor CD36 protein was conducted using nitrocellulose membrane-based SELEX. PCR initially prepared the random library and followed by in vitro transcription. The stringency of selection was increased by reducing the concentration of rhCD36 protein and incubation time while increasing the concentration of yeast tRNA. Introducing the BSA protein after five rounds of the SELEX cycle might increase the chances to isolate high affinity and specificity of RNA aptamer to rhCD36 protein. In our study, the counter SELEX with BSA protein was conducted at cycle 8th, 10th, and 12th. Table 1 shows the SELEX condition in this study and the optimized number of PCR cycles.
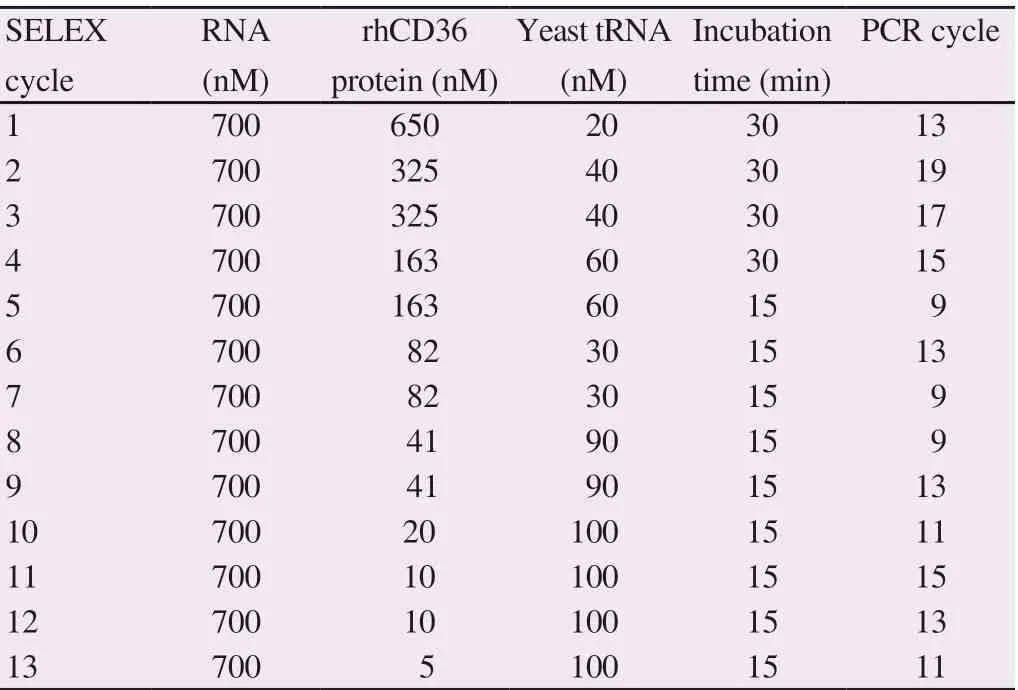
Table 1. Condition for SELEX experiment of RNA aptamers against rhCD36 and optimized number of PCR cycles.
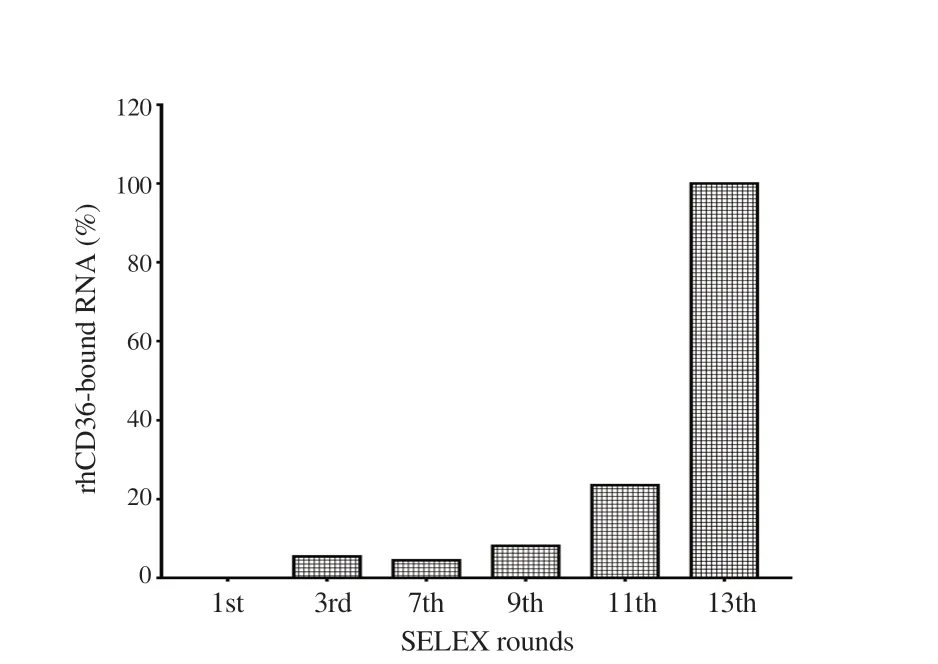
Figure 1. The SELEX enrichment of RNA pool targeting protein using semiquantitative PCR. The image represents the percentage of RNA that bound to rhCD36 from selected SELEX cycles. The ImageJ quantification shows that the RNA pool from 13th SELEX cycle demonstrated the highest percentage of bound RNA to rhCD36 protein.
3.2. Evaluation of SELEX enrichment
SELEX enrichment of RNA pool binding with rhCD36 was evaluated using sqRT-PCR at 1st, 3rd, 7th, 9th, 11th, and 13th SELEX cycle. Figure 1 shows the band intensity of amplified cDNA of collected RNA from selected SELEX cycle. The equal concentration of RNA from selected SELEX cycles and rhCD36 protein were used in the binding assay. The complexes were separated using nitrocellulose and reverse transcribed the bound RNA. Amplification of cDNA was limited to thirteen PCR cycle, and PCR products were loaded into the gel followed by ImageJ quantification (Supplementary Figure 1). The increasing recovery of bound RNA was observed starting at 3rd SELEX cycle and reached a maximum at 13th SELEX cycle.
3.3. Sequence and structure analysis of aptamer
After the 13th SELEX cycle, the amplified cDNA was cloned and sequenced. Sixty-six clones of rhCD36 aptamer candidates were successfully sequenced, aligned and classified into several groups based on their sequence frequency and homology. From Table 2, eleven clusters of aptamer sequence have been identified targeting rhCD36. Cluster 1 (15.15%), C1_RC60 showed the highest frequency of occurrence, which was 13.64%. However, C1_RC17 that had a single frequency of occurrence was grouped similar to C1_RC60 as it only showed a single mutation at position (43C>U) within the randomized region. There were four sequences of cluster 2 (7.58%) (C2_RC15, C2_RC04, C2_RC71, and C2_RC51) with single mutation at position (48U>C for C2_RC04 and 27A>G for C2_RC71) compared with C2_RC15, both present within randomized region. However, C2_RC15 showed a single base deletion at position 39delG. In cluster 3 (6.06%), there was one mutation site within randomized region identified in C3_RC57 that occurred at position 50U>C compared with C3_RC25. Other isolated clusters were C4_RC26 (6.06%), C5_RC16 (4.55%), C6_RC46 (4.55%), C7_RC14 (3.03%), C8_RC11 (3.03%), C9_RC52 (3.03%), C10_RC67 (3.03%) and C11_RC55 (3.03%). The aptamer clusters were also confirmed using Neighbor-Joining (NJ) tree (Supplementary Figure 2). Figure 2 shows the significant motif sequence found by MEME software was located within a randomized region (green) instead of two constant regions (red and light blue). The motif sequence of rhCD36 aptamer clusters was discovered at ‘[U/G/A][U/G]GG[A/U/G]GG[A/U/G]GG[A/U][G/U]G[A/U/G]’. Besides, the sequences of isolated RNA aptamers were analyzed using online QGRS Mapper in order to determine the location of the G-quadruplex sequence. We also discovered that all the isolated RNA aptamers possess G-quadruplex sequence within the randomized region (Supplementary Figure 3).
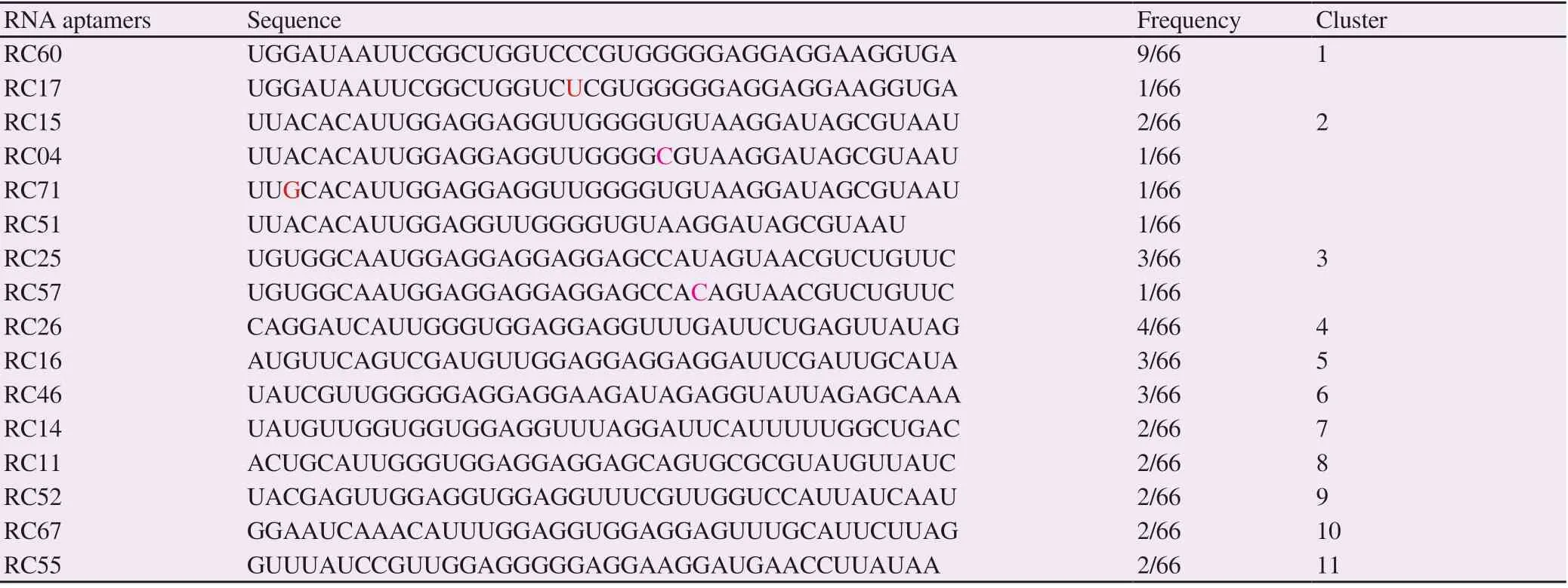
Table 2. The random sequence of isolated RNA aptamer clusters targeted rhCD36 protein according to the frequency number and degree of sequence similarities.
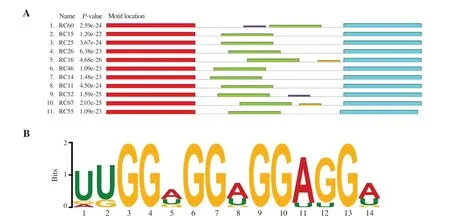
Figure 2. The sequence analysis of isolated aptamers by online MEME software. (A) Significant motif sequence was found within the randomized region (green) instead of constant sequence (red and light blue). The purple and yellow rectangle show other motifs found by software with insignificant E-value. (B) The thickness of a letter indicates the probability of base insertions between aptamer clusters.
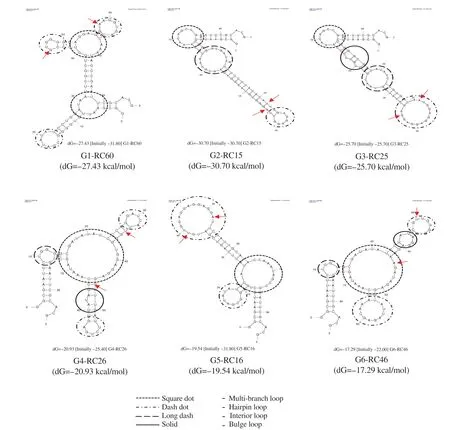
Figure 3. Secondary structure prediction of RNA aptamers that bind to rhCD36 protein using the online mfold software. The arrows indicate the discovered motif sequence.
3.4. Secondary structure prediction
The predicted secondary structure of 6 selected RNA aptamers represented for cluster 1-6 as showed in Figure 3. Most of the aptamer clusters had a hairpin loop structure connected to the multibranch or core loop. Also, there were also several clusters having bulge loop, and interior loop connected to core loop structure. The secondary structure analysis showed most of the motif sequences were located within a single-stranded of hairpin loop structure and double-stranded of the hairpin stem. Therefore, it can be hypothesized that the discovered motif sequence might play a role in the aptamer-target interactions as most of the motifs were located between hairpin loop structure and base pairs region within the randomized sequence.
3.5. Binding assay of isolated RNA aptamer
The binding assay was performed to confirm whether isolated RNA aptamers can bind to rhCD36 proteins. In this study, EMSA and sqRT-PCR were performed to confirm the aptamer binding. The interaction between isolated RNA aptamer with the target rhCD36 protein was observed on EMSA (Figure 4 and Supplementary Figure 4). Unfortunately, some of the complexes retained in the well, and this might be due to complexes aggregation. Statistical analysis of sqRT-PCR assay using unpaired t-test showed significant binding of isolated RNA aptamer compared with the RNA library (initial library) in nine aptamer candidates for targeted rhCD36 (Supplementary Figure 5). The result obtained from the sqRT-PCR assay was in concordance with the EMSA-based binding assay. Therefore, three aptamer candidates (RC60, RC04, and RC25) targeted rhCD36 proteins were selected to represent cluster 1 to cluster 3 and further evaluated for their binding affinity as they showed higher binding to rhCD36 compared to other aptamer clusters.
3.6. Dissociation constant (Kd) value of isolated aptamers
The Kd value was determined using a modified agarose-based EMSA assay. The concentration of RNA aptamer was fixed while the concentration of protein was varied (25 to 500 nM). Thus, the estimated Kd values for RC04, RC60 and RC25 were (17.49±4.16) nM, (21.90±2.40) nM, and (23.07±3.49) nM, respectively (Supplementary Figure 6-8). The results demonstrated that the binding affinity of RC04 had the highest binding affinity followed by RC60 and RC25.
3.7. The specificity of isolated RNA aptamer
In this study, ELONA was used to rapidly assess the relative binding specificity of several isolated RNA aptamers to the rhCD36, BSA and rhICAM-1. Previously, RC04 showed the highest binding affinity compared to RC60 and RC25. However, in the binding specificity analysis, RC04 candidate showed cross-reactivity to BSA and rhICAM-1 protein while RC60 and RC25 showed significantly no cross-reactivity to BSA and rhICAM-1 protein (Figure 5). Negative binding assay of RC60 and RC25 to nitrocellulose membrane filter also showed no binding (Supplementary Figure 9).
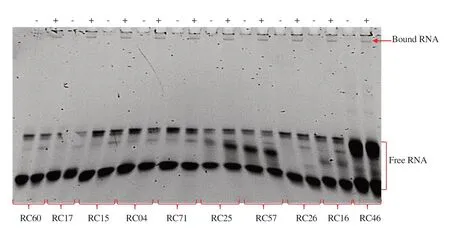
Figure 4. Electrophoretic mobility shift assay (EMSA) of selected RNA aptamers targeting rhCD36.
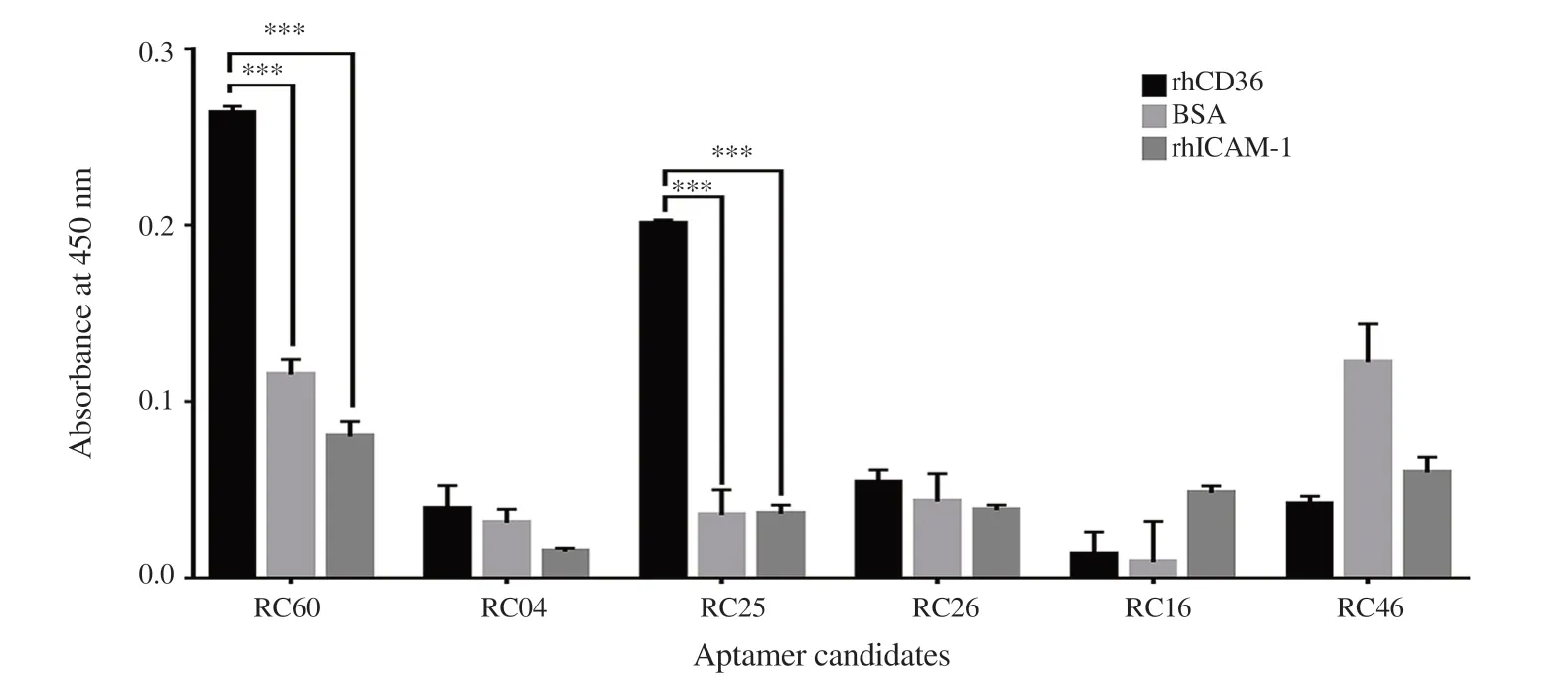
Figure 5. Binding specificity of isolated RNA aptamers to rhCD36. The binding specificity of isolated RNA aptamers was estimated by comparing the binding between rhCD36, BSA and rhICAM-1 protein. Data analysis was done using Two-way ANOVA (Multiple comparisons test) where ***P < 0.001. Error bar indicates ±SD of the triplicate experiment.
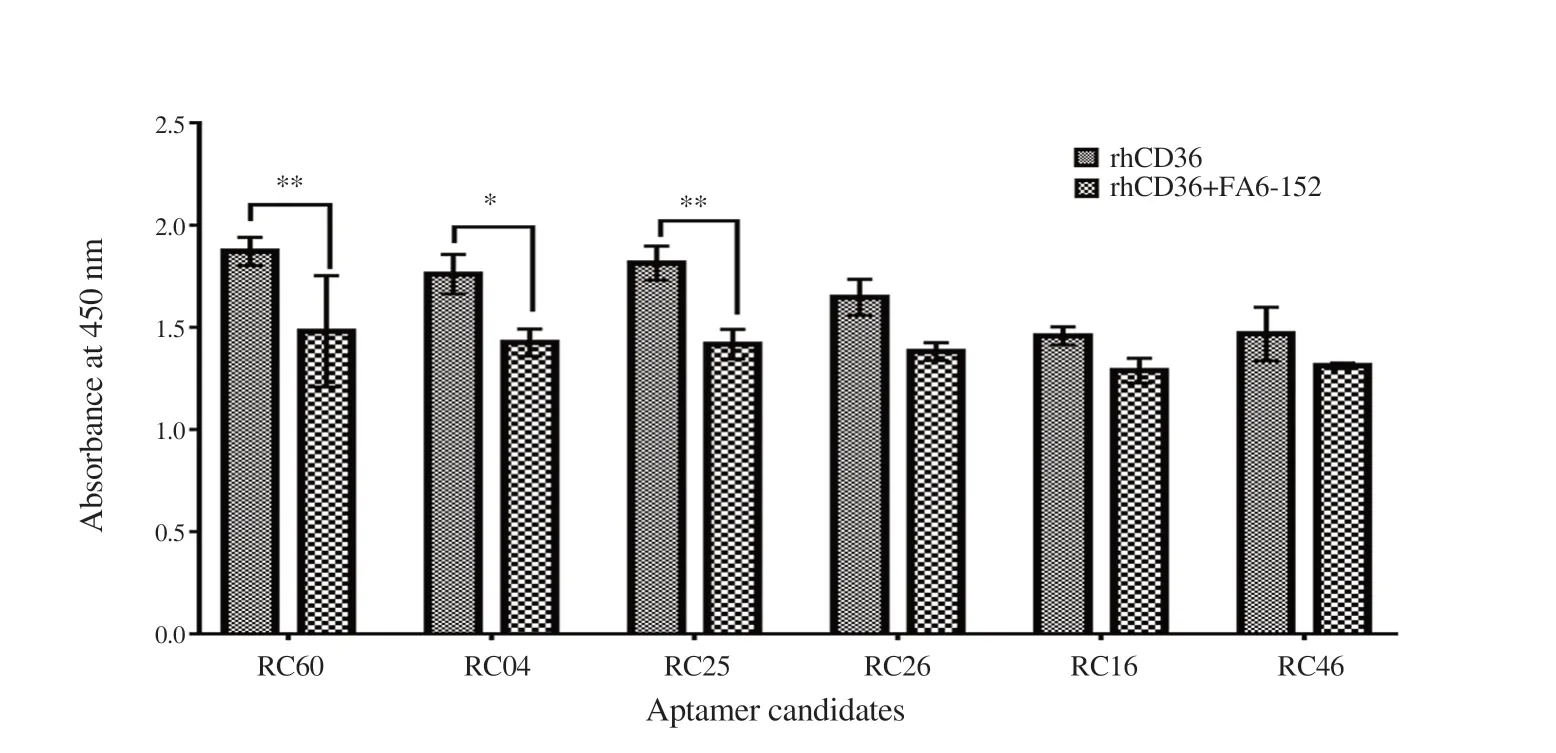
Figure 6. Enzyme-linked oligonucleotide assays (ELONA) for binding site confirmation. The polyA tail RNA aptamer (100 nM) was immobilized onto ELISA microplate. The competitive inhibition of aptamer binding was measured by quantifying the absorbance of bound rhCD36 in the presence of mAb FA6-152. The data obtained were compared with control (in the absence of mAb FA6-152). Data analysis was done using Two-way ANOVA (Multiple comparisons test) where *P < 0.05 and **P < 0.01. Error bar indicates ±SD of the triplicate experiment.
3.8. The binding orientation of isolated RNA aptamer to rhCD36
Aptamer competitive binding assay was designed in order to determine the binding epitope of RNA aptamer to rhCD36 protein. Significant reduction of absorbance was observed for RC60, RC04, and RC25 in the presence of mAb FA6-152 (Figure 6). The reduction of absorbance might be caused by the competitive inhibition of mAb FA6-152 at the same epitope site of RNA aptamer binding to rhCD36. Other RNA aptamers (RC26, RC16, and RC46) showed a reduction on the signal intensity with no significant difference. The most convincing hypothesis is that the immobilized RNA aptamer increases the captured protein when there are more free epitopes compared to protein immobilization.
4. Discussion
Previous studies have reported that there is an association between cytoadherence of IEs to the host endothelial surface receptor protein and parasite burden in the blood vessel. The parasite surface protein (PfEMP1) was reported to bind to several hosts endothelial surface protein including CD36 which linked to uncomplicated malaria, but it also can indirectly support the cytoadhesion of the IEs to the microvascular brain which leads to the development of severe malaria. Thus, this study was proposed to isolate and characterize a group of novel RNA aptamers that bind to CD36 protein as a potential malaria adjunct therapy. Nitrocellulose membrane-based SELEX was performed to isolate the RNA aptamers that specifically bind to recombinant human endothelial surface receptor CD36 protein. In each round of SELEX cycle, the amounts of specific binders are present at low copy number compared to a non-specific binder which then requires optimum PCR method to increase amplification efficiency to avoid amplification bias and minimize the loss of potential high-affinity binders. Over-amplification of PCR product hypothetically tends to cause DNA mispairing as oligonucleotide template containing an abundance of different sequences of the randomized region that can anneal with each other, leading to more round of SELEX cycle[24,25]. Therefore, to avoid PCR over-amplification throughout this SELEX experiment, the number of PCR cycles was optimized in every SELEX cycle in the increment of two PCR cycles until the expected band size appears[26]. Then, the scale-up of the DNA library for the next SELEX cycle was carried out using an optimized number of PCR cycle, followed by ethanol precipitation before subjected to in vitro transcription. The SELEX enrichment analysis showed that the increased amount of specific RNA bound to rhCD36 protein starting from round three and it reached maximum binding at around thirteen. The results obtained were in concordance with another study when they found increasing RNA or DNA recovery after 3 to 5 rounds of the SELEX[27].
The sequence analysis revealed a G-quadruplex sequence between 11 and 12 bases was found within all the isolated RNA aptamers. As predicted, the presence of potassium ion (K¬+) at 2.7 mM in the selection buffer has increased the chances of G-quadruplex formation that plays an important role in aptamer binding[28]. The aptamer that contains G-quadruplex was reported to be more thermodynamically and chemically stable, which increases stability within the nuclease environment of the bloodstream[29]. Some studies reported that the contact of the aptamer and the target occurs in the single-stranded region, but it also can happen in the basepaired region[30]. Nevertheless, further study is needed to confirm this hypothesis using RNase/DNase footprinting assay and in silico Aptamer Docking[31,32].
There were three aptamer candidates (RC60, RC04, and RC25) that show a significant binding with the rhCD36 protein. The RC04 aptamer shows the highest binding affinity compared to RC60 and RC25 even though it shows only one sequence frequency in the cluster. This finding correlates with another study which reported that the aptamer sequence frequency is a poor predictor of aptamer affinity. Thus, evaluation of the cycle-to-cycle enrichment of aptamer is a better predictor of binding compared to frequency determination in the final pool of the SELEX cycle[33]. Assuming the added target proteins were active, and the stoichiometric of the RNA aptamer and protein formation was at a similar ratio, the Kd was estimated[34].
The binding specificity analysis of RC60, RC04 and RC25 showed that RC60 and RC25 demonstrated significantly no cross-reactivity. Meanwhile, RC04 showed cross-reactivity against BSA and rhICAM-1 protein. The ICAM-1 protein is an immunoglobulin (Ig) superfamily member that is expressed on the surface of endothelial membrane cells and leucocytes encompassing five extracellular Iglike domain (D1-D5), a transmembrane domain and a cytoplasmic domain[35]. There is a relationship of high cytoadherence and cerebral malaria that associated with adhesion of IE to ICAM-1 protein[36,37]. Furthermore, the binding orientation analysis showed that RC60, RC04, and RC25 have the same binding epitope with mAb FA6-152. This monoclonal antibody was known to be able to inhibit the binding of ox-LDL and recognition of apoptotic neutrophils by residues 155-183[38]. Moreover, several studies have reported that mAb FA6-152 also efficiently inhibited the binding of infected erythrocyte to CD36[12].
In conclusion, this study successfully isolated and characterized two potential RNA aptamers (RC60 and RC25) that have affinity against rhCD36. These isolated RNA aptamer candidates could be further investigated on its future use as anti-cytoadherence for severe malaria adjunct therapy.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
This work is supported by an Exploratory Research Grant Scheme (ERGS; 203/CIPPM/6730106) and Higher Institution Centre of Excellent (HICoE; 311/CIPPM/4401005) from the Malaysian, Ministry of Higher Education and UniversitiSains Malaysia Fellowship Scheme.
Authors’ contributions
NAANK, JNAS, NFMZ and KMFM performed the experiment. NAANK and KMFM performed the data and statistical analysis, manuscript preparation and editing. Both NAANK and KMFM authors contributed to the final version of the manuscript. KMFM designed and supervised the project. All authors approved the final version for submission.
 Asian Pacific Journal of Tropical Biomedicine2020年1期
Asian Pacific Journal of Tropical Biomedicine2020年1期
- Asian Pacific Journal of Tropical Biomedicine的其它文章
- Hepatoprotection by dandelion (Taraxacum officinale) and mechanisms
- Renoprotective effect of umbelliferone in high-fat diet/streptozotocin-induced type 2 diabetic rats
- Raspberry ketone attenuates high-fat diet-induced obesity by improving metabolic homeostasis in rats
- Antioxidant and anti-melanogenic activities of ultrasonic extract from Stichopus japonicus
- Anti-microsporidial effect of thymoquinone on Encephalitozoon intestinalis infection in vitro