
基于支持向量机的癌细胞经典分泌蛋白与非经典分泌蛋白识别研究
2020-01-10 03:32:24余乐正柳凤娟李东海郭延芝李益洲
四川大学学报(自然科学版) 2020年1期
余乐正, 柳凤娟, 李东海, 郭延芝, 李益洲
(1. 贵州师范学院化学与材料学院, 贵阳 550018; 2. 四川大学化学学院, 成都 610065)
1 引 言
恶性肿瘤(癌症)是当今对人类健康和生命威胁最大的疾病之一,并已成为我国人口死亡的首要原因[1]. 由于具有发展速度快、侵袭性强、易转移复发、预后差等特点,大多数癌症在晚期才被发现,导致治疗难度大,死亡率极高.现代医学研究结果表明,癌症越早被发现,其治愈的几率就越高. 因此,实现对早期癌症的有效检测已成为治愈癌症、延长患者生命的关键[2]. 在癌症的发生发展过程中,肿瘤细胞会释放出一类反映癌症存在与生长的物质——肿瘤标志物. 肿瘤标志物可存在于血液、体液、细胞或组织中,主要包括RNA,DNA,蛋白质等生物活性分子[3]. 通过对该类物质的快速准确检测,可为判断是否患有癌症、癌症类别、癌症分期、预后效果等提供实验依据. 由于不同发展阶段、不同种类的癌细胞分泌出的蛋白质类型和表达水平不尽相同,近年来分泌蛋白已成为肿瘤标志物的主要来源之一[4-9]. 例如,甲胎蛋白(AFP)、α-L-岩藻糖苷酶(AFU)、高尔基体蛋白73(GP73)等已成为肝癌临床诊断的主要检测指标[10],前列腺特异性抗原(PSA)则是前列腺癌最重要的早期检测指标[11].
根据是否含有N端信号肽,分泌蛋白可简单分为经典分泌蛋白(CSPs)和非经典分泌蛋白(NCSPs)两大类[12]. 通过经典分泌途径与非经典分泌途径,蛋白质均可被释放到癌细胞外,并参与癌细胞的相关生理过程. 已有研究证实,不同种类的癌细胞可分泌出相同的蛋白质,且这些蛋白质的分泌主要依赖于非经典分泌途径[13]. 因此,对癌细胞非经典分泌蛋白进行系统深入的研究,可为寻找到不同种类癌症间通用的肿瘤标志物提供理论参考. 基于蛋白质序列信息和支持向量机(SVM)算法,通过严格的特征筛选,本文构建了一个二元分类器以快速准确地识别癌细胞非经典分泌蛋白. 对于测试集,本方法总的预测准确率为99.81%,表明本方法可作为一种辅助工具用于不同种类癌症间通用蛋白标志物的筛选.
2 材料与方法
2.1 材 料
本实验所用数据主要来自于人类癌症分泌蛋白质组数据库(HCSD)[14]. HCSD已收录13种癌症的分泌蛋白数据,如肝癌、肺癌、乳腺癌、前列腺癌、胃癌、结直肠癌、鼻咽癌、宫颈癌、胶质母细胞瘤、膀胱癌、胰腺癌、卵巢癌、淋巴瘤等. 从该数据库中共得到23 225条癌细胞分泌蛋白,包括5 263条CSPs与17 962条NCSPs. 此外,从前期工作中[8],收集到147条CSPs与102条NCSPs作为独立测试集.
2.2 建模方法
作为现今最流行的机器学习算法之一,支持向量机已被广泛应用于解决各种分类问题. 由于采用了结构风险最小化准则,并具有坚实的理论支撑,支持向量机可较好地处理小样本、高维度、非线性、局部极小点等问题[15]. 在前期各类分泌蛋白的识别研究中[16-18],支持向量机均表现出良好的应用效果,故本文也采用支持向量机来构建预测模型.
2.3 模型的性能评估参数
为客观准确地评估模型的实际预测性能,本文选取了以下4个评价参数:灵敏度(SE),特异性(SP),准确率(ACC)和马氏相关系数(MCC)[19].
(1)
(2)
(3)
MCC=
(4)
公式(1)~(4)中,TP为真阳性,即正样本被准确识别的数量;FP表示假阳性,即负样本被错误识别为正样本的数量;TN表示真阴性,即负样本被准确识别的数量;FN表示假阴性,即正样本被错误识别为负样本的数量.
3 实 验
3.1 训练集与测试集
为去除掉原始数据中冗余的序列信息,提高模型的稳定性,以相似度阈值为25%,利用CD-HIT Suite[20]对原始数据进行处理后,共得到761条CSPs和2 715条NCSPs. 随机提取其中的70%作为训练集,剩余的30%作为测试集[21],故训练集最终由533条CSPs和1 901条NCSPs组成,而测试集则包含228条CSPs及814条NCSPs.
3.2 特征提取与表征
除所用实验数据与建模方法外,特征筛选在蛋白质的分类预测研究中也发挥着非常重要的作用. 本研究分别采用氨基酸组成、自协方差变量、位置特异性得分矩阵以及信号肽来表征蛋白质中氨基酸的序列信息、邻接效应、进化信息及结构信息.
3.2.1 氨基酸组成 氨基酸组成(AAC)代表了20种常见氨基酸在蛋白质序列中出现的频率,每条蛋白质均被描述为一个20维的数字向量.
3.2.2 自协方差变量 在蛋白质的分类研究中,自互协方差(ACC)常用于计算蛋白质序列中氨基酸残基间的邻接效应. 自互协方差共包含两种变量,即相同描述符间产生的自协方差变量(AC)与不同描述符间形成的互协方差变量(CC). 由于自协方差变量的维数远小于互协方差变量的,且前者对邻接效应的贡献度远大于后者[22],故本文只采用自协方差变量来表征氨基酸残基间的邻接效应. 此外,前面的研究工作[23]已对自协方差变量的相关计算公式进行了详细描述,此处不再赘述. 由于本研究选用了疏水性、等电点、极性、转移自由能、侧链体积等5个理化性质,且氨基酸间的最大距离取值为5,故每条蛋白质最终被转化为一个25维的数字向量.
3.2.3 位置特异性得分矩阵 由于能有效表征蛋白质序列中氨基酸残基的进化信息[24],位置特异性得分矩阵(PSSM)已被广泛应用于各种蛋白质的分类研究. 利用PSI-BLAST程序(期望值阈值为10-3)对Swiss-Prot数据库进行搜索,并经3次迭代后,获得了每条蛋白质的位置特异性得分矩阵. 通过相关公式[23]对这些矩阵进行统一处理后,每条蛋白质均被转换为一个20维的数字向量.
3.2.4 信号肽 是否含有N端信号肽是经典分泌蛋白与非经典分泌蛋白结构间最显著的差异,故信号肽已成为区分两者的一个重要特征. 作为目前预测能力最强、应用范围最广的信号肽识别软件,SignalP 4.1[25]被用于蛋白质N端信号肽的识别,并通过D-score值予以表征.
3.3 蛋白质替代模型
基于上述特征,本文共建立了7个蛋白质替代模型:模型1仅含氨基酸组成(AAC);模型2仅含位置特异性得分矩阵(PSSM);模型3为氨基酸组成与自协方差变量融合形成的伪氨基酸组成(PseAAC);模型4为氨基酸组成与位置特异性得分矩阵融合形成的伪位置特异性得分矩阵(PsePSSM);模型5由氨基酸组成与信号肽融合而成;模型6由伪氨基酸组成与信号肽融合而成;模型7由伪位置特异性得分矩阵与信号肽融合而成.
3.4 模型的构建
本文最终的支持向量机预测模型是通过libsvm 3.12 (http://www.csie.ntu.edu.tw/~cjlin/libsvm/)工具箱建立起来的. 选择径向基函数(RBF)为模型核函数,并利用网格搜索法对模型的正则化参数C和核函数参数γ进行优化.此外,作为最客观的模型性能检测方法之一[26],留一法(Jackknife test)被用于构建最终的预测模型.
4 结果与讨论
4.1 特征筛选及替代模型的确定
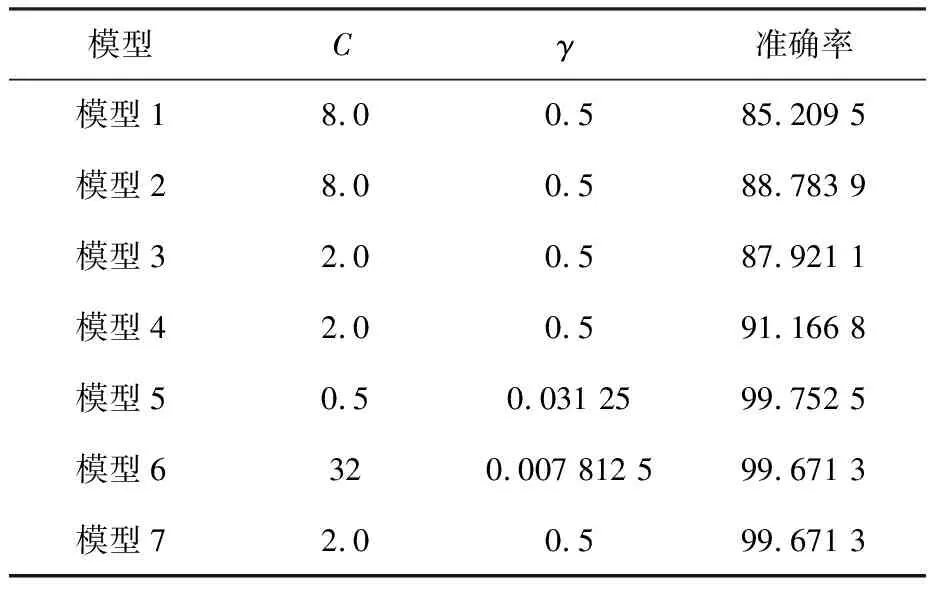
基于3.3节描述的7个蛋白质替代模型,本文共构建了7个支持向量机预测模型,相关训练结果均列于表1中.
表1不同蛋白质替代模型对训练结果的影响
Tab.1 Performance of different protein substitution models
模型Cγ准确率模型18.00.585.209 5模型28.00.588.783 9模型32.00.587.921 1模型42.00.591.166 8模型50.50.031 2599.752 5模型6320.007 812 599.671 3模型72.00.599.671 3
根据模型1与模型2的训练结果,PSSM对蛋白质的表征能力略优于AAC,表明PSSM的确能较好地反映蛋白质序列中氨基酸残基的进化信息. 模型3、模型4的训练结果表明,AC和PSSM的加入的确能有效提高模型的预测性能,且PSSM所包含的信息量多于AC的. 比较前4个模型与后3个模型的训练结果,信号肽的加入使得模型5~7的预测性能均有较大幅度的提升,表明信号肽在CSP与NCSP的分类研究中的确发挥着重要作用.同时,正是由于信号肽对CSP和NCSP过于强大的区分能力,使其掩盖了蛋白质替代模型PseAAC与PsePSSM之间的性能差异.虽然模型5的预测准确率最高,但模型7的优化参数最为合理,包含的信息量更多,且两者之间的预测准确率相差很小,故本文选择模型7作为最终的蛋白质替代模型.
4.2 模型的实际应用
利用3.1节构建的测试集,对模型5~7的实际预测性能进行了比较,相关测试结果均列于表2中.
表2不同SVM模型对测试集的预测结果
Tab.2 Prediction results of different SVM models obtained by analyzing the test sets

蛋白质类型CSPsNCSPs合计测试集数据2288141 042模型5准确预测数2288051 033准确率 (%)10098.9899.14模型6准确预测数2288071 035准确率 (%)10099.1499.33模型7准确预测数2268141 040准确率 (%)99.1210099.81
如表2所示,虽然模型5、模型6准确识别出所有228条CSPs,但它们对NCSPs的预测性能均弱于模型7. 模型7不仅准确识别出测试集中所有814条NCSPs,其对癌细胞分泌蛋白总的预测准确率与MCC值也最高(99.81%与99.44%),表明以模型7为最终的蛋白质替代模型是正确的.
为进一步比较模型5~7的实际预测性能,通过2.1节提到的独立测试集再次进行了检测. 模型5~7均准确识别出所有147条CSPs,且模型5将2条NCSPs错误预测为CSPs,而模型6和模型7仅错误预测1条NCSP. 进一步的研究发现,三个模型均错误预测的蛋白质(Q86UK5)在UniProt数据库中被标注为膜蛋白,SignalP 4.1也预测其为膜蛋白. 由于该蛋白质的D-score值为0.438,与SignalP 4.1的默认值(0.45)极为接近,这可能使得三个预测器均将其错误识别为CSP. 这一结果表明在区分经典分泌蛋白和非经典分泌蛋白时,还应注意区分分泌蛋白与膜蛋白.
5 结 论
经仔细分析癌细胞经典分泌蛋白与非经典分泌蛋白的各种特征,本文基于支持向量机算法构建了一个二元分类器以快速准确地识别癌细胞非经典分泌蛋白. 研究结果表明,本方法对癌细胞非经典分泌蛋白具有较好的预测性能,可作为一种辅助工具用于筛选不同种类癌症间通用的蛋白标志物.后续研究将尝试构建一个可快速准确区分不同种类癌细胞分泌蛋白的多元分类预测器,从而为寻找到每类癌症的特异性肿瘤标志物提供理论参考.
猜你喜欢
河北科技大学学报(2023年5期)2023-11-09 01:44:44
奥秘(创新大赛)(2019年9期)2019-10-09 02:03:56
小哥白尼(趣味科学)(2019年1期)2019-04-12 00:23:56
奥秘(2017年5期)2017-07-05 11:09:30
浙江农业学报(2017年3期)2017-04-08 02:39:02
自动化学报(2016年8期)2016-04-16 03:38:55
环境与生活(2016年6期)2016-02-27 13:47:10
无线电通信技术(2015年3期)2015-12-23 11:37:00
华东理工大学学报(自然科学版)(2015年4期)2015-12-01 04:00:23
中国科学技术大学学报(2013年8期)2013-03-11 20:18:37
