
长链非编码RNA HIT对急性淋巴细胞白血病SUP-B15细胞系伊马替尼抵抗的影响
2020-01-09 07:41:52史利欢刘俊闪王亚峰栗春香
中国药理学通报 2020年1期
史利欢,田 亮,黄 闪,刘俊闪,王亚峰,栗春香,刘 炜
[(郑州大学附属儿童医院,河南省儿童医院,郑州儿童医院(河南省小儿血液医学重点实验室),河南 郑州 450000)]
伊马替尼(imatinib,IM)作为酪氨酸激酶抑制剂(tyrosine kinase inhibitor,TKI),可通过竞争性与酪氨酸激酶结合,阻断三磷酸腺苷(adenosine triphosphate,ATP)对酪氨酸激酶的活化作用进而发挥抗肿瘤作用[1],其被广泛应用于治疗Ph(+)的急性淋巴细胞白血病(acute lymphoblastic leukemia,ALL)及慢性髓细胞白血病,可明显提高患者临床疗效及预后水平[2],但相当部分的患者会在5年内发生继发TKI耐药[3],严重影响治疗效果,因此明确IM耐药发生的机制对于Ph(+)的ALL患者的治疗具有重要意义。长链非编码RNA(long non-coding RNA,LncRNA)是一类转录长度>200nt的非编码RNA,在机体内发挥广泛且复杂的生物学作用,LncRNA HIT(HOXA transcript induced by TGFβ,HIT)近年被发现与多种肿瘤细胞恶性行为及进展相关[4-5],但其在ALL中的生物学作用尚不明确。本研究通过生物信息学及体外实验等多种手段对HIT与ALL IM抵抗的关系及相关机制进行了探究,情况如下。
1 材料与方法
1.1 细胞来源及培养人ALL细胞SUP-B15(美国ATCC,CRL-1929),人胚胎肾细胞293T(美国ATCC,CRL-3216),培养SUP-B15细胞采用10%胎牛血清(fatal bovine serum,FBS,美国Gibco)+ 100 U·mL-1青霉素(天津药业焦作有限公司)+100 μg·mL-1链霉素(国药集团国瑞药业有限公司)的 RPMI1640(美国Thermo Fisher Scientific)培养液;培养293T细胞采用10% FBS+100 U·mL-1青霉素+100 μg·mL-1链霉素的DMEM(美国Gibco)培养液。培养环境:37 ℃,5% CO2及100%相对湿度,SUP-B15细胞室温,1 000 rpm离心(fresco17,美国Thermo Fisher Scientific)5 min,每24 h更换培养液,原代细胞按1 ∶2分种传代。
1.2 SUP-B15 IM抵抗细胞株(IM resistance,IMR)及对照组(Control)细胞株的构建1 ∶2分种传代SUP-B15细胞,将细胞分为IMR及Control两组,IMR组细胞采用0.062 5、0.125、0.25、0.5、1、2 μmol·L-1IM(美国Selleck,S2475b)分步诱导,在每个IM浓度梯度下维持培养2周以上,待细胞生长状态良好,进入下一浓度的筛选,直至细胞在4 μmol·L-1IM中可维持生长,继续培养1周后完成IMR组细胞株的构建。同时采用等体积生理盐水处理SUP-B15细胞作为本研究的Control细胞株。
1.3 慢病毒转染敲降IMR细胞HIT的表达慢病毒包装表达靶向LncRNA HIT的shRNA 1#及2#载体(苏州吉玛基因公司),1 ∶2分种传代293T细胞,接种至6孔板中,分别编号为1#及2#两组,另取2支1.5 mL 离心管(德国Eppendorf)并编号,每管加入500 μL DMEM、1.5 μg Gag-Pol Rev expression vector(美国Addgene)及3.0 μg VSV-G expression vector(美国Addgene),并分别加入靶向LncRNA HIT的shRNA 1#及2#载体,充分混匀后室温静置5 min,加入转染试剂Lipofectamine 3000(美国Thermo Fisher Scientific)10 μL,充分混匀后室温静置20 min,PBS处理293T细胞3次,加入1 mL DMEM,缓慢均匀加入离心管中的混合液,6 h后PBS处理293T细胞3次,加入3 mL 10% FBS+DMEM培养液,48 h后收集上清液并过滤。将上清液与10% FBS+RPMI1640 1 ∶1混合后培养IMR细胞,连续培养3 d后,0.5 mg·L-1嘌呤霉素筛选细胞至其生长状态良好,完成IMR shHIT1#及2#两组细胞的构建。
1.4 CCK-8实验检测细胞IM半抑制浓度(50% inhibitory concentration,IC50)培养待检测对数生长期细胞,T20自动细胞计数仪(美国Bio-Rad)进行细胞计数,以1 000个细胞/孔接种待测细胞于96孔板中,细胞悬液总体积100 μL,设置0.062 5、0.125、0.25、0.5、1、2、4、8 μmol·L-1不同浓度的梯度IM,每组设置5个重复孔及空白对照孔,以相应浓度IM处理96孔板中的细胞24 h,每孔加入10 μL CCK-8试剂(日本同仁化学研究所),并充分混匀,37 ℃,5% CO2条件下孵育2 h,SynergyH1酶标仪(美国Biotek)检测各样本450 nm处吸光度,以空白孔调零,计算各孔的相对吸光度。
1.5 荧光定量PCR(fluorescence quantitative PCR)技术检测细胞中RNA表达水平收集对数生长期1×106左右的待检测细胞, 1 mL TRIzol试剂(美国Invitrogen)重悬细胞抽提细胞总RNA,Nanodrop2000超微量分光光度计(美国Thermo Scientific)检测RNA浓度,1 μg RNA使用逆转录试剂盒(美国Thermo Scientific Fermentas)逆转录为cDNA模板,配置合成LncRNA HIT、QKI、肿瘤干细胞(cancer stem cells,CSCs)标志物Oct4及Sox2基因qPCR引物(上海生工生物工程技术服务公司,详细序列情况见Tab 1)至工作浓度0.5 μmol·L-1,采用 SYBR Premix Ex Taq II核酸染料(日本TaKaRa)20 μL体系于Lightcycler-480Ⅱ荧光定量PCR仪(瑞士Roche)中进行qPCR反应,每组设置重复孔3个,GAPDH作为内参基因,2(-△△CT)法计算LncRNA HIT、QKI、Oct4及Sox2 mRNA相对表达水平。

Tab 1 The primer sequence of LncRNA HIT, QKI,
1.6 蛋白免疫印迹实验检测细胞中蛋白的表达水平收集对数生长期5×106左右的待检测细胞,500 μL Ripa蛋白裂解液(上海碧云天生物技术公司)4 ℃裂解细胞1 h,15 000 r·min-1,4 ℃离心细胞10 min,收集上清液于1.5 mL 离心管,BCA蛋白定量试剂盒(上海生工生物工程技术服务公司)检测总蛋白浓度,与聚丙烯酰胺凝胶电泳(polyacrylamide gel electrophoresis,PAGE)缓冲液配置成2 g·L-1体系,煮沸变性-20 ℃保存备用。配置15%聚丙烯酰胺凝胶,行PAGE,稳压120 V,转PVDF膜(美国Millipore)稳流300 mA,10%脱脂牛奶室温孵育PVDF膜2 h,剪取目的条带,1 ∶1 000的QKI抗体(anti-QKI,美国Abcam,ab126742);Oct4抗体(anti-Oct4,美国Abcam,ab18976);Sox2抗体(anti-Sox2,美国Abcam,ab97959);GAPDH抗体(anti-GAPDH,美国Abcam,ab70699)室温孵育目的蛋白分子量所对应的条带3 h,PBS洗条带3次后1 ∶2 000山羊抗兔IgG(sheep polyclonal to rabbit IgG H&L,美国Abcam,ab6795)室温孵育条带1 h,PBS洗条带3次后ECL化学发光底物(美国Thermo Pierce)孵育条带,ChemiDoc MP化学发光成像系统(美国Bio-Rad)对条带印迹进行曝光显影。

2 结果

Tab 2 Expression of LncRNA HIT, QKI, Oct4 and Sox2 mRNA in control, IMR, IMR shHIT1# and 2# cells
2.1 Control、IMR、IMR shHIT1#及2#细胞IM IC50的比较Control、IMR、IMR shHIT1#及2#细胞IM IC50分别为0.458±0.032、2.963±0.260、1.531±0.128及1.209±0.112 μmol·L-1,其中IMR、IMR shHIT1#及2#细胞IM IC50明显高于Control细胞(F=34.31,ControlvsIMR q1=13.89,ControlvsshHIT1# q2=7.37,ControlvsshHIT2# q3=4.16,P均<0.05),且IMR shHIT1#及2#细胞IM IC50明显低于IMR细胞(IMRvsshHIT1# q4=9.73,IMRvsshHIT2# q5=6.52,P均<0.05),见Fig 1。
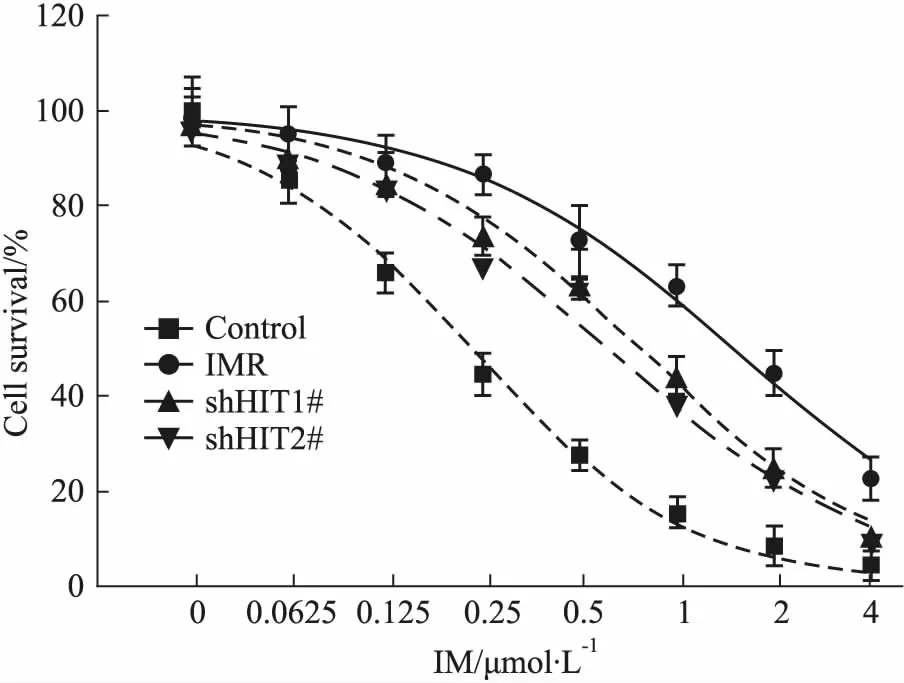
Fig 1 Comparison of IC50 to IM of control, IMR,IMR shHIT1# and 2# cells
2.2 Control、IMR、IMR shHIT1#及2#细胞LncRNA HIT、QKI、Oct4及Sox2 mRNA表达水平及比较Control、IMR、IMR shHIT1#及2#细胞LncRNA HIT、QKI、Oct4及Sox2 mRNA表达水平情况见Tab 2,统计学结果显示,IMR细胞LncRNA HIT表达水平明显高于Control细胞,IMR shHIT1#及2#细胞LncRNA HIT表达水平明显低于IMR及Control细胞(F=56.97,ControlvsIMR q1=12.95,ControlvsshHIT1# q2=4.63,ControlvsshHIT2# q3=6.32,IMR vs shHIT1# q4=14.12,IMRvsshHIT2# q5=16.33);IMR细胞、IMR shHIT1#及2#细胞QKI mRNA表达水平明显低于Control细胞,而IMR shHIT1#及2#细胞QKI mRNA表达水平与IMR细胞相比无明显变化(F=44.21,ControlvsIMR q1=13.17,ControlvsshHIT1# q2=11.52,ControlvsshHIT2# q3=13.84,IMRvsshHIT1# q4=2.38,IMRvsshHIT2# q5=1.71);IMR细胞、IMR shHIT1#及2#细胞Oct4 mRNA表达水平明显高于Control细胞,IMR shHIT1#及2#细胞Oct4 mRNA表达水平明显低于IMR细胞(F=30.42,ControlvsIMR q1=13.32,ControlvsshHIT1# q2=6.23,ControlvsshHIT2# q3=4.70,IMRvsshHIT1# q4=7.08,IMRvsshHIT2# q5=8.62);IMR细胞、IMR shHIT1#及2#细胞Sox2 mRNA表达水平明显高于Control细胞,IMR shHIT1#及2#细胞Sox2 mRNA表达水平明显低于IMR细胞(F=54.57,ControlvsIMR q1=17.47,ControlvsshHIT1# q2=5.00,ControlvsshHIT2# q3=5.97,IMR vs shHIT1# q4=12.47,IMRvsshHIT2# q5=11.50)。
2.3 Control、IMR、IMR shHIT1#及2#细胞QKI、Oct4及Sox2蛋白表达水平及比较Control、IMR、IMR shHIT1#及2#细胞Oct4及Sox2蛋白表达水平变化与mRNA水平一致,IMR细胞、IMR shHIT1#及2#细胞QKI蛋白表达水平明显低于Control细胞,而IMR shHIT1#及2#细胞QKI蛋白表达水平明显高于IMR细胞,见Fig 2。
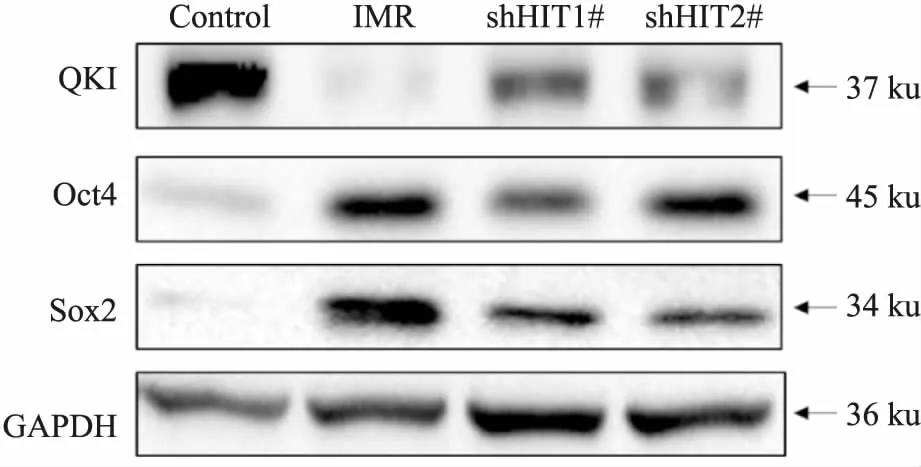
Fig 2 Expression of QKI, Oct4 and Sox2 protein in control,IMR, IMR shHIT1# and 2# cells
3 讨论
LncRNA HIT基因定位于HOXA基因簇中,受转录生长因子-β(transforming growth factor-β,TGF-β)的明显调控,在胚胎未分化的肢体、器官等组织中表达,并参与哺乳动物的软骨分化[6],近年许多研究指出,HIT在多种恶性肿瘤中表达重新上调,并参与其发生发展:Richards等[7]在研究中发现LncRNA HIT参与TGF-β诱导的乳腺癌上皮间充质转化过程(epithelial-mesenchymal transition,EMT),敲除HIT的表达后,TGF-β介导的小鼠乳腺癌细胞NMuMG侵袭、迁移能力均明显下调,提示HIT在乳腺癌的EMT调节和侵袭转移中发挥重要作用;Jia等[4]发现,HIT在非小细胞肺癌(non-small cell lung cancer,NSCLC)组织及细胞系中高表达,且HIT表达水平与患者总生存时间负相关,与患者临床分级及远处转移率正相关,体外通过慢病毒转染法构建过表达及shRNA敲降HIT的A549及SK-MES-1细胞模型,发现HIT可通过维持ZEB-1蛋白的稳定,促进NSCLC细胞侵袭转移及EMT;Yu等[5]发现,LncRNA HIT过表达或敲降可明显增加或减少NSCLC细胞增殖能力,同时明确其与转录因子E2F1的相互作用,进而对下游靶基因调控,发挥促进乳腺癌增殖的作用;李铸鹏等[8]的研究显示,LncRNA HIT在侵袭性乳腺癌组织中表达明显升高,且与早期NSCLC患者复发及预后相关,表明了HIT与NSCLC耐药性及发生发展具有潜在联系。但目前有关HIT在血液肿瘤中的生物学研究仍是空白,而在前期研究中,我们通过生物信息学(http://rbpdb.ccbr.utoronto.ca/)分析发现LncRNA HIT与抑癌因子RNA结合蛋白Quaking(QKI)存在潜在的相互结合,暗示了HIT可能通过调控QKI在血液肿瘤中发挥作用。
QKI近年被许多研究报道可作为重要的抑癌因子影响多种恶性肿瘤的发生发展:Zong等[9]研究发现,QKI在肺癌组织中表达下调,并与患者预后时间相关,体内外实验发现过表达QKI可抑制肺癌细胞的恶性增殖,其作用机制与阻滞Notch信号通路的激活有关;Zhao等[10]研究显示,QKI在前列腺癌组织中表达明显降低,且其表达与肿瘤细胞分化程度、患者TNM分期及总生存率密切相关,体内外实验发现QKI可抑制前列腺癌细胞的增殖及成瘤能力;He等[11]发现QKI在结直肠癌组织及细胞系中表达下调,且受到微小RNA-155(MicroRNA-155)的负相调控,在结直肠癌中发挥抑制细胞周期和侵袭的作用;Lu等[12]研究发现QKI在口腔癌组织及口腔癌干细胞系中低表达,体外及裸鼠移植瘤实验结果显示QKI可通过特异性结合Sox2基因3'UTR区域阻滞Sox2的转录,进而抑制口腔癌细胞CSCs特性及恶性表型。QKI同样被发现与血液肿瘤的发生发展具有潜在联系:Fang等[13]发现QKI是红细胞成熟的关键分子,提示QKI对造血干细胞的正常分化具有重要意义,同时,Tili等[14]发现慢性B型淋巴细胞性白血病患者肿瘤细胞中QKI表达水平明显下调,且通过转基因小鼠模型发现QKI的表达与白血病的进展明显负相关。而CSCs则可通过原癌信号通路的激活及抗凋亡等多种途径促进恶性肿瘤细胞包括IM在内的多种药物抵抗的发生[15]。由此我们提出假设:HIT是否可通过调控QKI的表达进而影响ALL细胞CSCs表型发挥生物学作用。
本研究在IM抵抗Ph(+)的ALL细胞系中发现LncRNA HIT表达的上调,提示HIT可能参与ALL细胞IM抵抗的发生,而采用shRNA下调IM抵抗细胞中HIT的表达后,细胞IM IC50明显降低,表明HIT对ALL细胞IM抵抗具有促进作用,但shHIT1#及2#细胞IM IC50仍高于Control细胞,推测是由于ALL细胞IM抵抗的发生可能涉及多条信号通路,而HIT仅作为其中重要的一部分参与IM抵抗的发生。为了进一步明确HIT发挥生物学作用的相关机制,我们对可能的相互作用分子QKI的表达进行了检测,发现IM抵抗的ALL细胞中QKI mRNA及蛋白表达水平均下调,提示QKI表达的下调与ALL细胞IM抵抗的发生密切相关,结合生物学分析结果,我们推测HIT可能与QKI存在相互作用,共同影响ALL细胞的IM抵抗,因此,我们在shHIT1#及2#细胞中检测了QKI的表达,发现敲降HIT后,QKI mRNA表达水平无变化,而蛋白表达升高,表明HIT可通过负调控QKI蛋白表达水平,影响ALL细胞IM抵抗。基于QKI与肿瘤干细胞标志物表达的紧密关系,我们对相关分子Oct4及Sox2表达水平进行了检测,发现在IM抵抗的ALL细胞中Oct4及Sox2 mRNA及蛋白表达均明显上调,提示IM抵抗与ALL细胞干细胞特性密切相关,而敲降HIT后Oct4及Sox2 mRNA及蛋白表达下调,表明HIT对于Oct4及Sox2的表达存在正调控作用。
综上,我们的研究表明HIT可通过抑制ALL细胞QKI蛋白水平的表达,上调CSCs相关分子Oct4及Sox2,进而促进细胞IM抵抗能力。接下来我们将进一步探究HIT与QKI蛋白的相互结合及调控机制,并采用细胞及动物模型明确HIT在ALL中的生物学效应,为相关诊断试剂及靶向药物在血液肿瘤中的开发及应用提供新的标志物。
(致谢:本实验在河南省小儿血液医学重点实验室完成,感谢马平、李晶、陈静、谢昕对实验的指导及帮助)。
猜你喜欢
中老年保健(2021年5期)2021-08-24 07:06:20
小雪花·成长指南(2021年2期)2021-05-20 09:14:00
天津医科大学学报(2019年3期)2019-08-13 06:53:00
初中生世界·九年级(2019年4期)2019-05-05 01:07:12
中成药(2017年8期)2017-11-22 03:19:00
系统工程与电子技术(2016年2期)2016-04-16 05:16:53
中国现代医学杂志(2015年26期)2015-12-23 11:04:22
医学研究杂志(2015年7期)2015-06-22 11:01:42
中国光学(2015年1期)2015-06-06 18:30:20
西安交通大学学报(医学版)(2015年2期)2015-02-28 17:59:21
