
电感耦合等离子体发射光谱法同时测定地质样品中的钍和氧化钾
2019-12-31 06:10秦晓丽田贵李朝长蒋智林
岩矿测试 2019年6期
秦晓丽, 田贵, 李朝长, 蒋智林
(安徽省核工业勘查技术总院, 安徽 芜湖 241000)
钍燃料比铀燃料循环周期更长,在热中子反应堆中易获得更高的转化比,产生的核废料放射性低,而且钍储量比铀储量大3倍以上,是一种前景非常可观的能源材料[1]。天然存在的钾同位素有39K、40K、41K,其中只有40K(比活度为31.5Bq/g)具有放射性[2]。在地质找矿方面,铀、钍、钾的分布特征能够在一定程度上反映放射性矿产及伴生矿产资源的分布特性;在辐射环境方面,天然钍和40K的含量是评价电离辐射对人体产生内照射和外照射的两个必测参数,因此钍和钾的同时测量对于放射性矿产资源的勘探具有积极意义,也能为天然放射性生态环境评价提供重要依据。
地质样品的化学成分复杂,消解困难,且钍、钾含量差异很大,不同岩石中的钍含量均值一般在1.3~138.9μg/g,钾含量均值一般在0.3%~6.5%[3],钍、钾分别采用不同的仪器测定。目前EJ 349.3—1997《岩石中微量钍的分析方法》是地质样品中分析微量钍的行业标准,采用碱熔法对试样进行分解,阳离子交换树脂分离后用偶氮胂Ⅲ分光光度法测定钍[4],该方法的缺点是碱熔法会引入大量基体,前处理过程冗长、复杂且不利于多元素同时测定;而氧化钾是根据GB/T 14506.11—2010《硅酸盐岩石化学分析方法》用火焰原子吸收分光光度法测试,由于火焰原子吸收线性范围窄,浓度高的溶液需要稀释,测定结果误差较大,且效率较低[5-6]。
电感耦合等离子体发射光谱法(ICP-OES)的线性范围宽、稳定性高,适合多元素的同时测定,是目前较为普遍而经济的分析手段,已被广泛应用于稀有多金属矿[7]、水系沉积物[8-9]、土壤[10-12]、长石[13]等地质样品的分析。近年来已有采用ICP-OES法测定钍的相关报道,用过氧化钠碱熔分解试样以后再过滤分离、洗涤、溶解沉淀,可以准确测定地球化学样品中的钍[14-15]。这种方法即使洗涤以后可以降低基体干扰,但是操作过程繁琐,也不利于多元素的同时测定。用ICP-OES法单独测定氧化钾,分析成本较高,大都是与其他常量元素一起列入待测项目[13,16]。目前尚没有常量元素氧化钾和微量元素钍共同测定的相关报道。
本文尝试采用硝酸、氢氟酸和高氯酸消解样品后,根据待测元素的含量高低配制适合的曲线范围,用ICP-OES法同时测定其中的钍和氧化钾。探讨了溶样过程中溶剂的选择,选择了待测元素的最佳分析谱线以减少光谱干扰,用国家一级标准物质验证了本方法的可行性。
1 实验部分
1.1 仪器及工作条件
Avio200型电感耦合等离子体发射光谱仪(美国PerkinElmer公司),工作条件为:射频发生器(RF)功率1500W;等离子体流量8L/min;辅助气流量0.2L/min;雾化气流量0.7L/min;观测方式为径向观测;读数延迟时间30s。
1.2 主要试剂
实验用水为屈臣氏纯净水;硝酸、氢氟酸、盐酸及高氯酸均为分析纯。
1.3 标准曲线的配制
1.3.1钍和氧化钾标准储备液的配制
钍标准储备液的配制(1000μg/mL):依据EJ 349.3—1997。准确称取2.3790g优级纯硝酸钍,置于200mL烧杯中,加入20mL硝酸加热至其完全溶解,冷却后转入1000mL容量瓶中,加纯水到刻度,摇匀。经标定获得钍标准溶液的质量浓度,再配制不同质量浓度的钍标准工作液。
氧化钾标准储备液的配制(1000μg/mL):依据GB/T 14506.11—2010。准确称取500~600℃灼烧2h的高纯氯化钾1.5829g于烧杯中,加纯水溶解后,移至1000mL容量瓶中,加入10mL硝酸,加纯水至刻度,摇匀。
1.3.2钍和氧化钾标准工作液的配制
钍标准中间溶液(10μg/mL、1μg/mL):准确吸取1000μg/mL钍标准储备液5mL于500mL容量瓶中,再加入10mL 50%的硝酸,加纯水至刻度(此介质为1%的硝酸),其浓度为10μg/mL;准确吸取10μg/mL钍标准储备液10mL于100mL容量瓶中,再加入2mL 50%的硝酸,加纯水至刻度(此介质为1%的硝酸),其浓度为1μg/mL。
钍及氧化钾标准系列浓度:分别吸取一定量的标准中间溶液,定容至刻度,介质为1%的硝酸,配成系列标准曲线,钍的浓度(μg/mL)为:0、0.010、0.025、0.050、0.100、0.250、0.500;氧化钾的浓度(μg/mL)为:0、10、25、50、100、200、300、400。
1.4 样品分解及测定
称取约0.15g样品于100mL聚四氟乙烯烧杯中,少量水润湿,加入3mL硝酸摇匀,再加入10mL氢氟酸和2mL高氯酸,摇匀,加聚四氟乙烯烧杯盖,然后将其置于110℃调温电热板上消解2h左右,升温至130℃保持2h,开盖后继续升温至160℃加热2h(为了达到良好的挥硅效果,隔20~30min摇晃一次),继续升温至200~230℃,加热至高氯酸白烟冒尽,取下冷却后加入0.5mL 50%的硝酸,少量纯净水冲洗烧杯壁,置于调温电热板170℃加热,微热10min至溶液清亮,取下冷却后将溶液转移至25mL容量瓶中,用纯水定容至刻度,摇匀,用ICP-OES进行测定。
将仪器性能调至最佳分析状态,以金属浓度为横坐标,发射强度为纵坐标,绘制标准曲线,再测试试剂空白和样品,若样品含量超过标准曲线的范围,可以适当稀释后再重新测试。
2 结果与讨论
2.1 溶样试剂的选择
地质样品化学成分复杂,碱熔法虽能溶解大部分样品,但所带的基体杂质多且不利于多元素的同时测定,一般采用多种酸混合消解的方法,如盐酸-硝酸-氢氟酸-高氯酸[17]。地质样品尤其是土壤中存在大量的硅,一部分金属元素存在于土壤的矿物晶格中,且晶格比较稳定,只有用氢氟酸才能破坏这类晶格[10],但氢氟酸对某些盐类的溶解性很差,为保证样品溶解完全,一般会与硝酸混合使用。此外,地质样品中还存在有机质,加入高氯酸或硫酸等高沸点且氧化性强的酸分解试样,一方面可以除去样品中的有机质,另一方面由于蒸发冒烟时所需的温度高,剩余氢氟酸易除尽。但由于硫酸的沸点(338℃)特别高,很难被除去且易跳溅,一般不选择用硫酸。
本文选择较有代表性的水系沉积物标准物质GBW07309、GBW07312为参考样品进行试验,这两种标准物质含一定量的有机质,二氧化硅含量(64.89%~77.29%)适中。用盐酸-硝酸-氢氟酸-高氯酸(方法1)和硝酸-氢氟酸-高氯酸(方法2)两种体系消解样品,其中方法1中,先加入盐酸后放在电热板上蒸干,取下冷却后再加入硝酸-氢氟酸-高氯酸。由表1分析结果可知两种溶样体系对分析结果无明显差异,但方法1比方法2增加了盐酸的消耗,浪费试剂、污染环境且样品处理过程复杂,故最终本方法采用硝酸-氢氟酸-高氯酸消解样品,盐类残渣易分解,效果较好。
表1不同混合酸消解样品中钍和氧化钾的测定结果比较
Table 1 Comparison of analytical results of Th and K2O in samples dissolutied with HCl-HNO3-HF-HClO4and HNO3-HF-HClO4acid digestion

样品处理方法GBW07309GBW07312Th(μg/g)K2O(%)Th(μg/g)K2O(%)盐酸-硝酸-氢氟酸-高氯酸(方法1)14.132.0022.392.87硝酸-氢氟酸-高氯酸(方法2)14.321.97 23.262.91认定值12.4±0.71.99±0.0621.4±1.12.91±0.04
2.2 分析谱线的选择
分析谱线的选择直接影响到测试结果的准确性,待测元素分析谱线的选择必须考虑其背景干扰、灵敏度等因素。由于地质样品成分复杂,离子种类繁多,它们对含量低、灵敏度差的痕量元素干扰十分严重。对此类干扰,可通过选择干扰少的分析谱线来减少。
本实验对每个元素选择强度适中、背景轮廓适合的分析谱线进行多次测试,由表2测定结果可知,当钍的测定波长为401.913nm,氧化钾的测定波长为766.490nm时,测定结果的相对误差小于13.0%,相对标准偏差(RSD)小于4.0%,满足规范要求。
表2钍和氧化钾的最佳分析谱线
Table 2 Analytical spectral lines for thorium and potassium oxide determination

测定次数GBW04120GBW04121Th(μg/g)K2O(%)Th(μg/g)K2O(%)283.730nm401.913nm766.490nm283.730nm401.913nm766.490nm111.08 3.82 0.9343.1124.636.19210.00 3.92 0.9552.8423.596.17312.103.700.9048.5323.146.15测定平均值11.06 3.81 0.93 48.1623.796.17认定值 3.4±0.10.84 21.7±0.56.08相对误差(%)225.2912.1610.32121.949.621.48RSD(%)9.492.892.7210.123.210.32相关系数0.99930.99990.99980.99930.99990.9998
2.3 方法技术指标
2.3.1检出限
按照消解样品的方法制备10份试剂空白溶液,在仪器最佳稳定状态下测定试剂空白值,钍的空白值(μg/mL)为:0.002、0.002、0.003、0.006、0.003、0.003、0.004、0.006、0.003、0.003;氧化钾的空白值(μg/mL)为:0.540、0.136、0.336、0.577、0.423、0.302、0.201、0.278、0.103、0.151。以空白值标准偏差的三倍计算得出钍、氧化钾的方法检出限分别为0.69μg/g、0.008%(称样量为0.1500g,定容体积为25mL)。
汪君等[15]用ICP-OES法测定了地球化学样品中的钍,该项研究采用碱熔法处理样品,分离过滤沉淀后再进行酸溶,钍的检出限为0.21μg/g。本方法检出限比汪君等[15]高,但溶样简单且有利于多元素的同时测定。DZ/T 0130—2006《地质矿产实验室测试质量管理规范》关于地球化学调查样品分析方法中水系沉积物、土壤试样分析质量控制要求是:钍、氧化钾的检出限分别为2μg/g、0.05%,由此可知本方法能满足大多地质样品的分析要求。
本方法的不足之处在于,对特别低含量的钍和氧化钾结果不够满意,其原因可能是方法检出限偏高,需要检出限更低的仪器或更合适的样品处理方法来检测。
2.3.2准确度和精密度
选取土壤和水系沉积物两种不同岩性的4个国家一级标准物质(GBW07409、GBW07404、GBW07310、GBW07302a),每个标准物质称取6份,按照消解样品的方法分解试样,用ICP-OES测定,其准确度及精密度结果见表3。由表3可知本方法的测定平均值与该标准物质的认定值基本吻合,测定平均值与认定值的对数误差的绝对值(lgC)均在0.1以内,表明该方法准确、可靠,可应用于实际分析检测中。钍、氧化钾的相对标准偏差(RSD)均小于6.0%,优于汪君等[15]的方法(RSD为7.7%~15.9%)。本方法精密度好,离散程度小,重复性好。
表3准确度和精密度测定结果
Table 3 Accuracy and precision tests of the method

2.3.3加标回收率
为了进一步验证方法的准确性,本文进行了加标回收实验。从10μg/mL钍标准溶液中分别取0、0.15、0.20、0.25、0.30mL标准溶液,在1000μg/mL氧化钾标准溶液中取0、2.0、3.0、3.5、4.0mL标准溶液,加入国家标准物质GBW07304a,按样品消解的方法分解试样,用ICP-OES测定,加标回收率为96.0%~104.0%(表4),结果较满意。
表4加标回收率试验
Table 4 Spiked recovery tests of the method
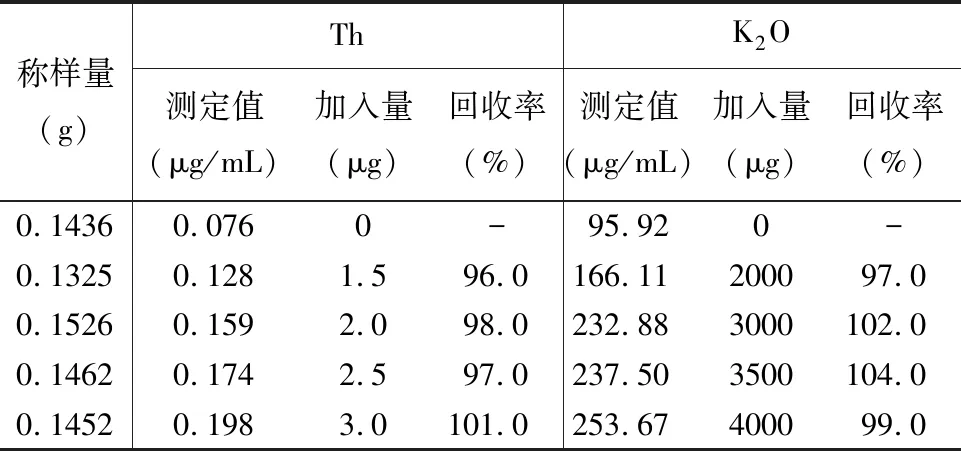
称样量(g)ThK2O测定值(μg/mL)加入量(μg)回收率(%)测定值(μg/mL)加入量(μg)回收率(%)0.14360.0760-95.920-0.13250.1281.596.0166.11200097.00.15260.1592.098.0232.883000102.00.14620.1742.597.0237.503500104.00.14520.1983.0101.0253.67400099.0
3 结论
用硝酸、氢氟酸、高氯酸溶解样品,硝酸提取,ICP-OES法分别在波长401.913nm和766.490nm处,采用径向观测方式测定地质样品中的钍、氧化钾,其标准曲线线性良好,方法检出限、准确度、精密度和加标回收率都符合DZ/T 0130—2006《地质矿产实验室测试质量管理规范》的要求。
由于采用酸溶法处理样品,避免了碱熔法带来的基体干扰,并且可以在不增加分析成本的情况下,分取此溶液用激光测铀仪同时进行铀元素的分析测试,提高了效率,适合于大部分地质样品中钍、氧化钾含量的同时测定。该方法已应用于实际样品测试,成图效果良好。
猜你喜欢
天津化工(2022年2期)2022-04-26
中学生数理化·高一版(2020年11期)2020-12-14
中学化学(2019年2期)2019-07-08
发明与创新·中学生(2019年6期)2019-06-26
科学与财富(2018年26期)2018-10-24
中国科技纵横(2018年10期)2018-07-27
浙江化工(2018年1期)2018-02-03
中国资源综合利用(2016年6期)2016-01-22
舰船电子工程(2012年8期)2012-07-11
数理化学习·高一二版(2009年5期)2009-07-31
