
SRA数据库架构及二代测序数据共享
2019-12-27 07:25:02李瑞华田国祥郭晓娟李豹张军吕军
中国循证心血管医学杂志 2019年11期
李瑞华,田国祥,郭晓娟,李豹,张军,吕军,5
以454测序技术、Solexa基因组分析技术、SOLiD测序技术及Thermo Sciences/Ion Torrent半导体芯片测序技术为主流的二代测序技术的出现意味着高通量测序的实现[1,2],人类进行一系列基因组水平的研究被带入了一个更高层次。大规模的基因组研究和高通量测序技术的不断发展,使生物数据面临井喷式增长[3],由于二代测序技术可同时对大量短片段测序,这些海量测序数据将具极其复杂性及高通量性,使得存储传统测序数据的一些数据库如Trace Archives数据库等不能适应新的测序结果。鉴于此,在2007年底,NCBI(National Center for Biotechnology Information)推出SRA数据库,主要用来储存、显示、下载、分析及共享二代测序数据。SRA数据库建立伊始,名为Short Read Archive,后来改为Sequence Read Archive[4]。SRA建立至今,各项测序数据数量急剧上升(图1),目前已成为美国国立卫生研究院(NIH)存储二代测序数据的主要数据库[5],同时是国际核苷序列联合数据库(INSDC)的一部分,可与欧洲生物信息学中心(EBI)和日本DNA数据库(DDBJ)之间进行数据共享。
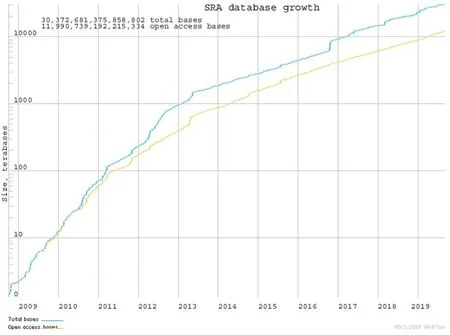
图1 SRA数据库收录数据增长曲线
1 SRA数据库的数据组织架构
1.1 Meta数据指与测序实验及其实验样品相关的数据,如实验目的、实验设计、测序平台、样本数据等等,Meta数据又包含以下层次:
①S t u d y——研究课题 s t u d y 的检索号(accession number)以前缀DRP,ERP或SRP开头。study是就实验目标而言的,一个study包含一个或多个experiment。
②Sample——样本信息 sample的检索号以前缀DRS,ERS或SRS开头。sample可以包括物种信息、菌株(品系)信息、家系信息、表型数据、临床数据,组织类型等。
③Experiment——实验信息 experiment的检索号以前缀DRX,ERX或SRX开头。experiment是SRA数据库的最基本单元,对一个或多个样本进行测序,产生的测序数据以runs的形式存储于SRA[4]。大多数描述性信息都是在SRA experiment级别捕获的,并将显示在公共记录中,提交者须为每个experiment提供清晰且信息丰富的标题和说明。
1.2 序列数据包括序列及其质量信息等,在SRA数据库中以run为单元存储。run的检索号以前缀DRR,ERR或SRR开头。一个实验可以包含一个或多个run。
2 SRA数据库的基本使用
进入SRA数据库官网:https://www.ncbi.nlm.nih.gov/sra,点击SRA Toolkit Documentation(图2)。
在SRA Toolkit Documentation页面选择SRA Toolkit Installation and Configuration Guide(图3),打开提供的下载链接(图4),找到与电脑操作系统相对应的下载工具安装包(图5),进行SRA Toolkit下载,下载成功后将压缩包解压,解压后可见一bin文件夹,各种测序下载工具和多种的数据格式转换工具即在该文件夹内,其中“prefetch”是常用的数据下载工具,“fastqdump”是常用的fastq格式转换工具(图6)。
在S R A 数据库首页搜索框内输入相关研究,可以是疾病名称或者序列数据等(本文以“肺癌lung cancer”为例),或点击搜索框下的“advanced”进入高级检索界面,通过限定词进行更精确的检索(图7),SRA提供了“OR、AND、NOT”即“或与非”几个操作以达到更精确方便的查找。
通过在SRA数据库搜索“lung cancer”,结果显示目前有17 714个数据集,每个数据集链接下均提供相应的SRA ID,界面右侧还显示对应物种的数据集个数(图8),点击每一个数据集链接均可进入相应详细信息界面。
选择打开任一个数据集链接,即可得到其详细信息界面,以第一个数据集为例,打开后显示该数据集不同层次数据的项目编码(图9)。点击study项目编码,得到该study的详细信息(图10),显示这个研究的实验数、run数及数据量,本例中study包含15个experiments和15个runs。
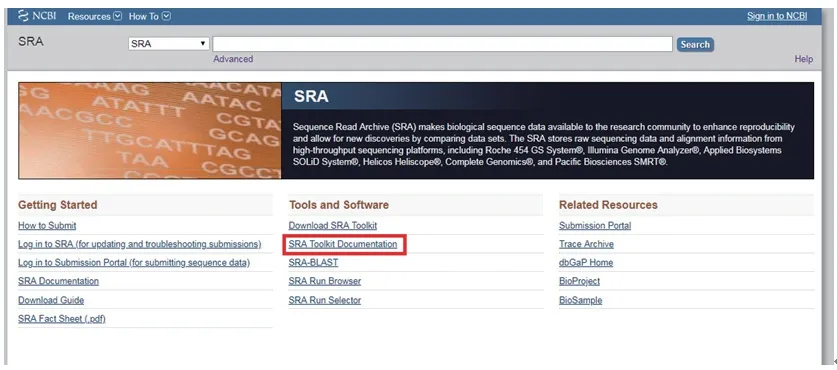
图2 SRA数据库官方网站首页
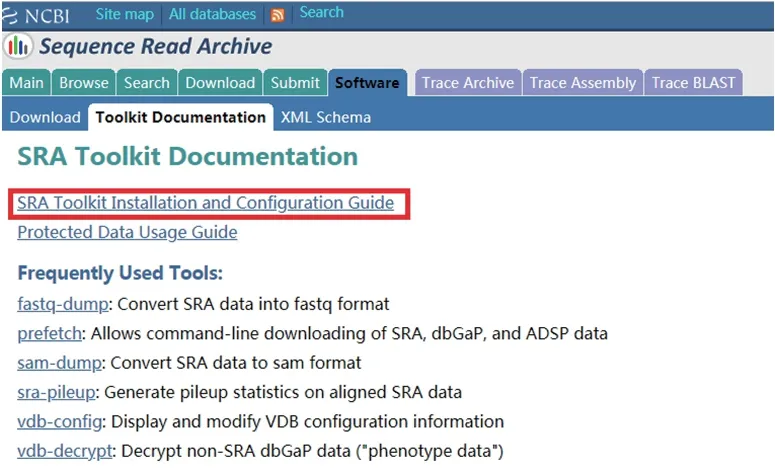
图3 SRA Toolkit Documentation页面
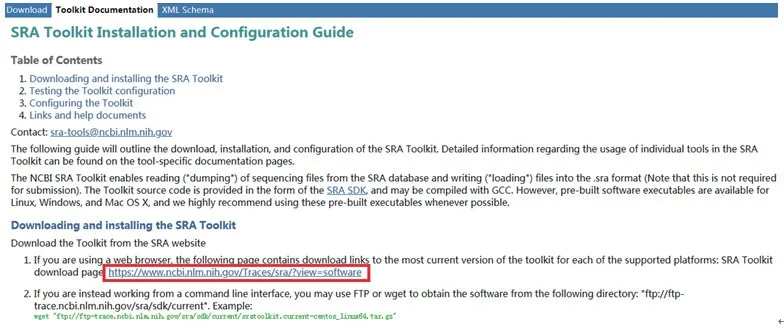
图4 SRA Toolkit下载链接
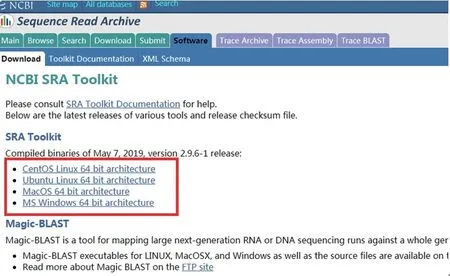
图5 下载SRA Toolkit安装包
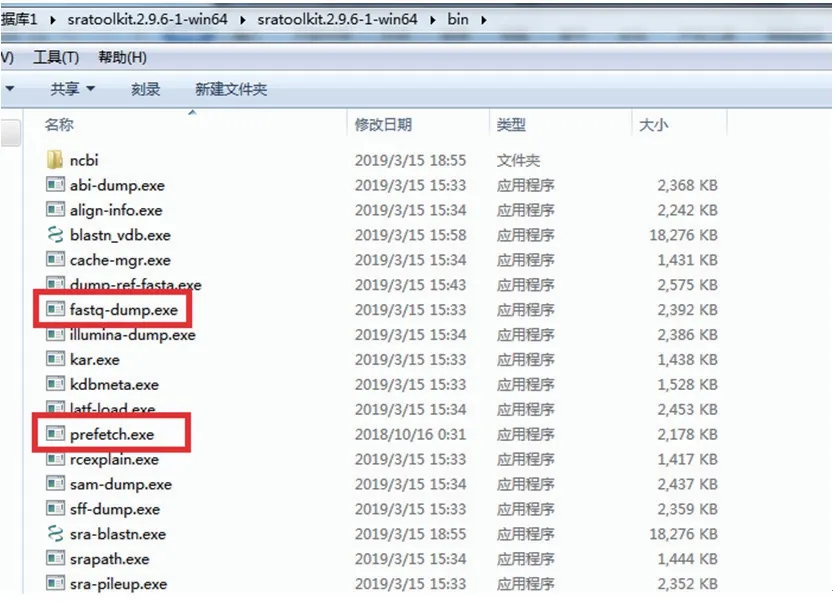
图6 SRA Toolkit内bin文件夹下各项工具
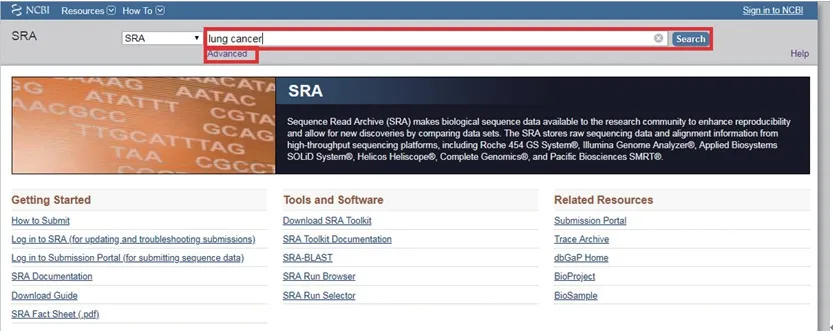
图7 SRA数据库首页进行检索
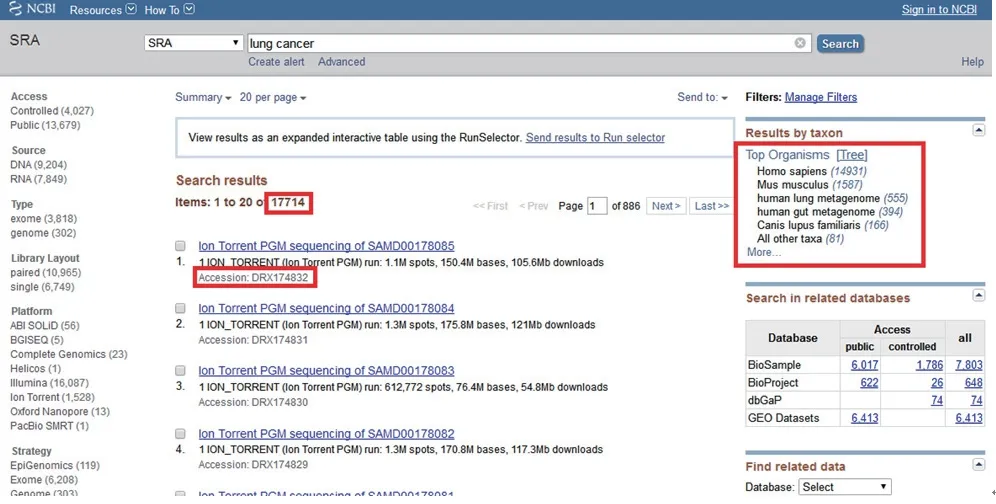
图8 搜索研究疾病所得数据集
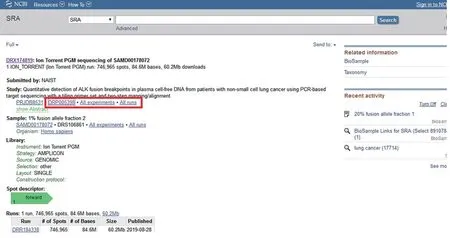
图9 数据集详细信息

图10 study详细信息
分别点击图9中All experiments及All runs(或点击图10中显示的实验数15及run数15),可依次得到experiments及runs的详细信息(图11~12)。
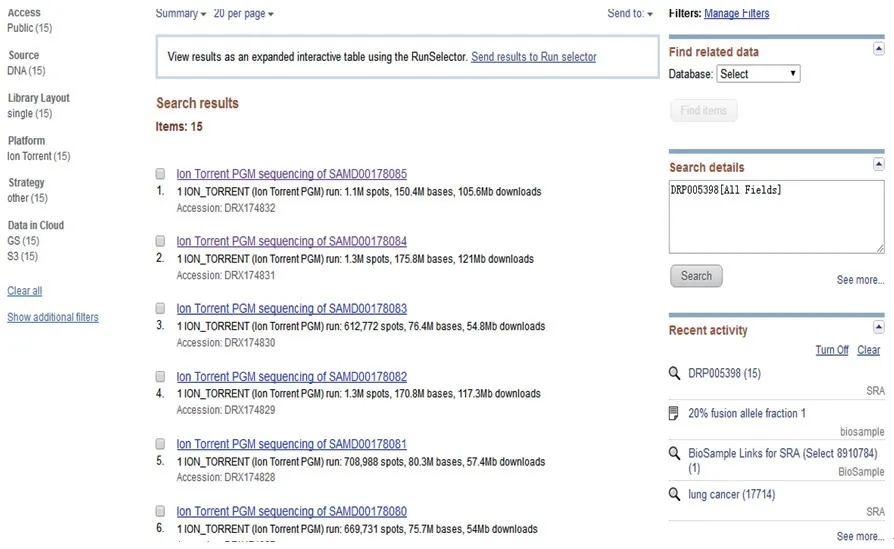
图11 experiments的详细信息
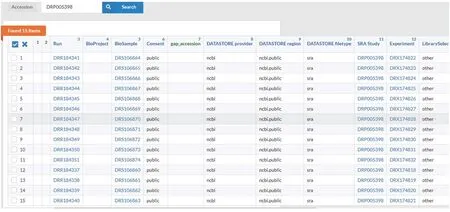
图12 runs的详细信息
图13 下载安装aspera connect
3 SRA数据库序列数据下载
从SRA数据库下载高通量的序列数据,可直接在SRA数据库网页下载或者利用上文提到的SRA Toolkit下载,但两种方法均耗时过长,此时可采用一种大数据下载工具Aspera。首先下载Aspera:到Aspera网站(https://downloads.asperasoft.com/en/downloads/8?list)下载操作系统对应的aspera connect,进行安装(图13)。
安装完成后,将其安装路径下的bin目录添加到环境变量中,具体步骤是:首先复制该文件所在的路径,打开控制面板→“系统”(或系统与安全)→“高级系统设置”,点击右下角的“环境变量”,在“环境变量”界面“系统变量”中选择“Path”,点击编辑,将所复制路径粘贴到变量值后方,注意路径之间要用分号隔开,点击全部的“确定”键保存,环境变量即设置成功。
打开计算机命令提示符(方法有很多,这里介绍一种:按“win+r”键打开运行面板,输入“cmd”,点击“确定”),使用下方命令进行下载所需数据:
ascp -v -k 1 -T -l 200m -i "C:UsersAdministratorAppDataLocalProgramsAsperaAspera Connectetcasperaweb_id_dsa.putty"dbtest@sra-download.ncbi.nlm.nih.gov:data/sracloud/traces/dra2/DRR/000001/DRR001472 ./
上述命令中C:UsersAdministratorAppDataLocalProgramsAsperaAspera Connect是aspera connect的安装路径,traces/dra2/DRR/000001/DRR001472是所需数据的路径,可根据实际需要进行更改,本文以“DRR001472”为例进行介绍,路径获取方式如下图(图14)(直接在图14中点击右下方红色框内链接也可直接下载,但下载速度常较慢)。运行上述命令即可得到DRR001472数据文件(图15)。
4 将原始数据转换为fastq格式
由于适用于大部分生物软件的是fastq格式数据,所以我们需要将下载的原始数据转换为fastq格式。具体方法是:打开命令运行界面,输入以下内容并运行:fastq-dump DRR001472,即可进行格式转换(图16),转换完成后,fastq格式数据存在于原始数据相同目录下(图17)。
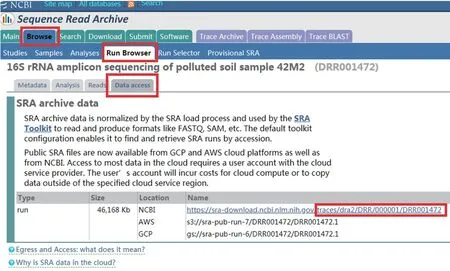
图14 DRR001472下载路径获取
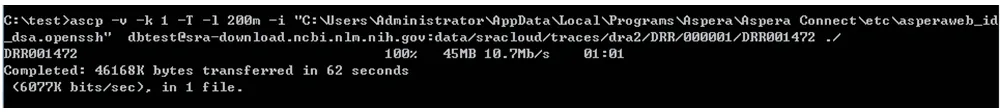
图15 DRR001472序列下载成功

图16 fastq格式数据转换
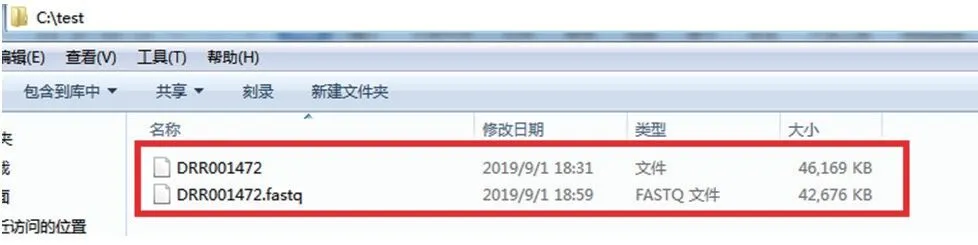
图17 fastq格式数据转换
5 总结
随着大数据时代降临,呈爆炸式井喷式激增的海量数据资源在各个领域开始量化进程[6]。数据的可再利用性、数据共享政策正引起全球普遍重视[7]。SRA数据库作为存储二代测序原始数据的代表性数据库,接受来自各种测序项目数据[8],对于广大生物信息学研究者提供了极具前景及研究价值的生物分析平台。本文从SRA数据库架构、数据下载及数据格式转换方面等进行相关介绍, 旨在减少科研工作者在数据获取、软件使用方面所花费的时间和精力,提高科研效率。
猜你喜欢
新民周刊(2022年27期)2022-08-01 07:04:49
传染病信息(2021年6期)2021-02-12 01:52:58
解放军医院管理杂志(2020年5期)2020-02-14 17:00:40
疯狂英语·爱英语(2020年9期)2020-01-07 00:52:42
意林图解作文(小学版)(2019年6期)2019-07-16 08:35:46
安徽农业科学(2018年32期)2018-05-14 08:59:54
专利代理(2016年1期)2016-05-17 06:14:36
求学·理科版(2016年3期)2016-03-23 05:46:24
生物医学工程学进展(2015年1期)2015-02-28 14:53:42
化学工业与工程(2015年1期)2015-02-10 03:01:41
