
基体合金元素对SiC/Al界面结合影响的第一性原理及实验研究
2019-12-16 08:32:14邹爱华周贤良康志兵饶有海吴开阳
无机材料学报 2019年11期
邹爱华, 周贤良, 康志兵, 饶有海, 吴开阳
基体合金元素对SiC/Al界面结合影响的第一性原理及实验研究
邹爱华, 周贤良, 康志兵, 饶有海, 吴开阳
(南昌航空大学 材料科学与工程学院, 南昌 330063)
采用基于密度泛函理论的第一性原理及实验相结合的方法, 探讨了Al基体中分别掺杂Mg、Si、Cu合金元素对SiC/Al界面结合的影响, 重点考察了合金元素在界面偏聚时的电子结构和成键情况。研究表明: 在未掺杂Al/SiC体系界面结构优化时, 以Si原子为终止面的Brigde结构是最稳定的结合方式; 当合金元素分别替换界面处的Al原子后, 界面处原子的分波态密度、Muliken电荷及成键原子集居数等电子结构参数均有不同程度的变化, 这不仅增加了界面处 Si与Al原子结合, 同时也增强了界面处和亚界面处的Al基体和SiC增强相原子之间的相互作用, 使体系更加稳定, 界面黏着功均有不同提升; 其中掺Mg提升效果最明显, 其次为掺Cu和掺Si; 利用第一性原理计算的掺杂Al/SiC体系黏着功和实验值较为接近且变化规律相同。
合金元素; SiC/Al界面; 界面结合; 第一性原理
SiCP/Al复合材料由于具有较高的比刚度、比强度、耐磨性、热导率以及较低膨胀系数等优异性能, 在航空航天、汽车制造以及电子封装材料领域有着广泛的应用[1-4]。界面是复合材料的重要组成部分, 是基体与增强体载荷传递的“枢纽”, 对复合材料的力学性能和物理性能有着极为重要的影响[5-8]。然而SiC/Al复合材料界面属于非润湿体系, 当制备温度较低时, 两者很难润湿, 导致界面结合差; 当温度升高时, 虽然有利于改善界面间的结合, 但界面处却容易形成有害界面反应产物, 如Al4C3等脆性相, 而且易水解, 这将严重影响复合材料的力学或耐蚀性能[9-10]。
在Al基体中添加合金元素是提高SiCp/Al体系润湿性的重要方法之一, 添加合金元素主要是通过以下两种机制来改善金属/陶瓷体系的润湿性[11]: 1)液体或固体表面富集, 以降低液态金属表面张力或固、液界面张力; 2)在界面发生化学反应, 生成界面反应产物。目前国内外许多学者针对基体合金化处理并探讨其对复合材料界面润湿的影响, 开展了一系列的研究工作。如Laurent等[12]研究了在973~ 1173 K温度范围内添加5wt%~12wt% Si对SiC/Al体系润湿性的影响, 发现Si几乎很少改进体系的润湿性, 但随着Si含量的增加, 能够抑制界面反应并可小幅提高润湿性。Choh等[13]则发现Si在1173~ 1373 K温度范围内能够提高润湿性, 认为Si元素的存在能够缩短润湿潜伏期。Ferro等[14]也提出Si能提高SiC/Al体系润湿性, 并且当Si含量在Al-Si共晶点附近时润湿性最好。因而有关Si对SiC/Al体系润湿性的影响并不系统, 结论不一。马晓春等[15]讨论了含Cu元素对SiC/Al体系润湿的影响, 发现Cu会阻碍Al2O3氧化膜的消失, 从而降低SiC/Al体系的润湿性。张强等[16]采用预氧化处理SiC颗粒为增强体, 以含Mg铝合金为基体, 制备SiCp/Al复合材料, 发现材料制备过程存在界面反应, 生成一定数量的MgAl2O4, 从而提高了润湿性。刘俊友等[17]同样也以Mg含量不同的铝合金作为基体, 以氧化态SiC作为增强体制备复合材料, 发现当Mg含量为2wt%时, 复合材料界面会出现八面体尖晶石。当Mg含量大于4wt%时, 界面会发生反应Mg+SiO2→2MgO+Si, 生成致密纳米尺度的MgO, 反之形成MgAl2O4。这些针对不同基体合金元素对金属–陶瓷复合材料润湿性影响的研究未形成系统, 而且研究手段基本为传统的实验方法。
考虑到在实际金属和陶瓷润湿实验过程中, 影响因素较多, 如金属容易氧化、炉内气氛、陶瓷粗糙度等, 特别是基体中添加合金元素后, 元素在界面的偏聚或形成界面反应生成新相等, 实验所包含的影响因素实际极为复杂, 这样的传统研究方法难以从原子尺度上揭示界面结合的机理。
本研究拟采用基于密度泛函理论的第一原理计算方法, 综合考察基体合金元素(Mg、Si、Cu)在界面偏聚时对SiC/Al界面结合的影响; 以掺杂前后界面的电子结构信息分析界面的结合机理; 结合改良座滴法实验测量了其润湿角, 对用第一性原理模拟的结果作了验证。
1 材料与方法
实验用于测量润湿性的金属熔滴分别为99.99%纯铝, Al-Mg(5wt%)、Al-Si(5wt%)及l-Cu(5wt%)合金。金属合金材料均在氩气气氛下电弧熔炼而成。实验前将纯铝及其合金打磨成表面光滑, 重量约为50 mg的球型块体。实验中碳化硅采用-SiC陶瓷基板。考虑到无压烧结的-SiC表面会有一层SiO2薄膜[18], 会严重影响润湿角测量的准确性, 实验前研磨去除了基板上的氧化膜, 降低基板表面粗糙度。首先在金相抛光机上依次采用400#、800#和1500#金刚石磨盘对基板进行研磨, 然后依次使用20、10、5和0.5 μm不同粒度金刚石研磨膏抛光至镜面, 所得基板的具体参数如表1所示。最后将制成的铝合金和SiC基板放置在丙酮溶液中超声波清洗三遍以去除污渍。
实验中测试润湿性采用高温高真空润湿系统。该系统的工作原理为: 实验前用机械泵和分子泵相结合的方式使炉体保持真空状态, 通过加热电源、钨铼热电偶和PID温度装置控温, 采用高纯度金属Ta片加热; 为了保证系统的保温性,炉壁采用内层两层, Mo片加上外层四层不锈钢, 通过滴落系统和升降系统让铝滴到达SiC基板上进行润湿测试。润湿实验后, 利用ADSA软件将CCD数码相机拍摄的图像矢量化, 获取液滴的轮廓数据点, 最后采用SESDROPD软件对液滴进行分析拟合计算, 获取润湿数据。
2 计算模型建立
2.1 计算方法
本实验计算采用Materials Studio软件中的CASTEP[19](Cambiedge Serial Total Energy Package)软件包, 选择广义梯度近似(Generalized Gradient Approximation, GGA)下的PBE(Perdew-Burke-Ernzerhof)势函数用来描述关联作用。平面波截断能取值340 eV, 确保能量收敛到1×10–5eV/atom(1 eV/atom= 9.6485×104J/mol)。布里渊区用Monkhorst-Pack k点网格取样, 体相计算时k点网格选取8×8×8, 界面和表面的计算选择8×8×1。其他的参数均设为精确。几何优化时, 弛豫到每个原子间的力小于0.3 eV/nm,每个原子位移小于 0.0001 nm。为了加快收敛速度, 拖尾参数设置为0.01 eV。本研究采用超软赝势来描述离子与价电子之间的相互作用, 为了得到稳定精确的计算结果, 先对各晶胞的结构进行优化, 基于优化模型, 再构建界面模型进行计算。

表1 SiC基板的基本参数
2.2 模型的建立
选取Al(100)、(110)、(111)、(211)四个晶面以及4H-SiC(001)、(011)、(111)、(211)四个晶面来构建表面结构模型, 模型原子层的厚度均控制在0.8~1.0 nm, 真空层设置为1 nm, 先对结构模型进行优化, 然后计算体系能量。各表面的表面能计算结果如表2所示。Al(111)与SiC(001)表面能最低, 因而在构建两相界面时, 优先选择这两个面作为结合界面来构建SiC/Al界面模型。
为了进一步考虑计算的准确性及经济性, 分别对其Al及4H-SiC进行了表面收敛性计算。本研究根据SiC薄膜表面原子弛豫收敛, 确定SiC(001)表面厚度: 增加SiC(0001)表面原子层数, 如果薄膜表面原子层间距的弛豫量和上一个薄膜对应位置原子层弛豫变化量相同, 且中间原子层间的弛豫变化量几乎为零, 则认定薄膜是收敛的。其中分别对SiC(0001)-C和SiC(0001)-Si表面构型结构进行优化,并对原子层间距变化及其占体材料层间距百分比进行表征计算。优化弛豫计算表明: 当层数≥11时, SiC(0001)薄膜原子层间距变化量趋近收敛, 且中心层原子弛豫量几乎为零。
对于Al薄膜, 由于上下面只有一种原子, Al(111)面为非极性表面, 因此Al(111)薄膜可根据表面能的收敛性确定厚度, 计算发现当=4时, Al(111)薄膜表面能为0.86 J/m2, 随着层数的增加, 表面能几乎保持不变, 趋近于收敛。所以综合考虑计算的经济性和准确性, 选取SiC为11层、Al为4层。
SiC表面堆积有两种形式, 即界面处分别以Si原子和C原子为终端。以Si原子为终端的原子结构为例, 如图1所示。根据Al原子与SiC界面处原子的相对位置, Al/SiC界面结合主要有三种形式: Al原子在界面两个原子中间上方(A, bridge), Al原子直接在界面原子上方(B, top)和Al原子在四个原子中心位置上方(C, hollow)。
本研究选取Al(111)和SiC(001)两个低指数面构建模型, 经过计算此界面模型失配度仅为8.415×10–6, 根据Hong等[20]的计算, 在界面处失配度很小的情况下, 界面处产生应变很小, 可忽略不计, 可认为界面是完全匹配的。
2.3 SiC/Al界面结构优化
考虑到SiC(001)面有以Si为终止面和以C为终止面的两种构型, 本研究为确定两种构型的稳定性, 计算了其表面能随化学势的变化规律。表面能计算公式:
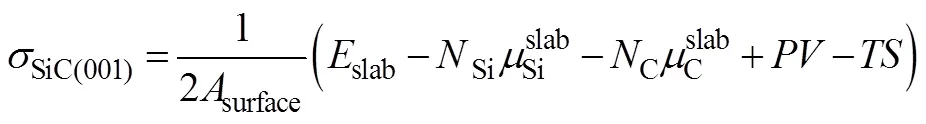
式(1)中: 为SiC(001)表面积; 为构建超晶胞SiC(001)薄膜优化后的总能量; NSi和NC分别为学势; P为压强, V为体积, T为温度, S则为体系的熵。在恒定压力下和0 K下, PV和TS值都为零, 忽略不计。经公式(1)的简化推导计算, 得出SiC(001)薄膜表面能和之间存在如图2的变化关系。

表2 Al以及4H-SiC晶体表面的表面能
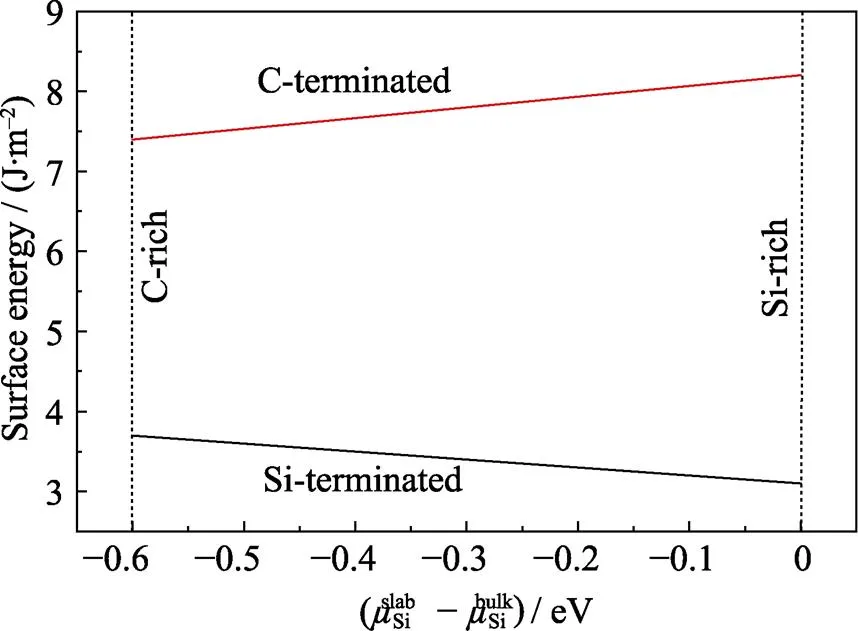
图2 SiC(001)表面能随变化的关系图
从图2中可以看出, SiC(001)表面能并不是一个固定值, 随着Si的化学势的改变而改变。在整个Si原子化学势变化范围内, 以C为终止面的表面能始终大于以Si为终止面的表面能。当表面能越大时, 表面越不稳定, 结构优化时会通过弛豫和重构来减小表面能。从能量角度看, SiC(001)表面更倾向于以Si原子为终止面。
另外比较计算了以C为终止面及Si为终止面的界面粘着功(ad)。ad通常用来表征界面结合强度, 粘着功越大则界面结合强度越强。根据UBER方法[21]计算SiC/Al界面分离功与界面间距的关系, 以确定平衡界面间距, 计算粘着功的公式为:
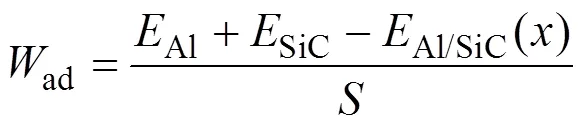
式(2)中:Al和SiC分别为SiC/Al界面中两侧Al和SiC薄膜原子结构优化后的能量;Al/SiC()表示为SiC/Al界面结构优化后的能量;表示SiC/Al界面的界面面积。
图3为分离功与界面间距的关系曲线。从图3可以看出, 以C为终止面的粘着功大于以Si为终止面的粘着功, 这与它们的表面能相一致, 表面能越大, 表面的活性越大, 表面原子越易与其他原子成键。界面间距对界面结合强度有着重要影响, 六种结构在界面间距为0.1~0.3 nm之间达到平衡位置, 约为0.2 nm, 粘着功最大; 随着界面间距的增大, 体系粘着功逐渐减小, 最终收敛趋近于0。虽然从总体上来看C终止界面的分离功大于Si终止界面, 但以Si为终止面的bridge-site (A-site)结构, 其粘着功的数值最大为1.43 J/m2。
该值与文献[22]报道的实验测试1173 K熔融A1在SiC表面的接触角约75°, 界面粘着功为1.04 J/m2较为接近。而且该值与本研究后续的润湿角实验测试及相应的粘着功相吻合。这说明当以Si为终止面的bridge-site结构是SiC/Al结合的主要方式。
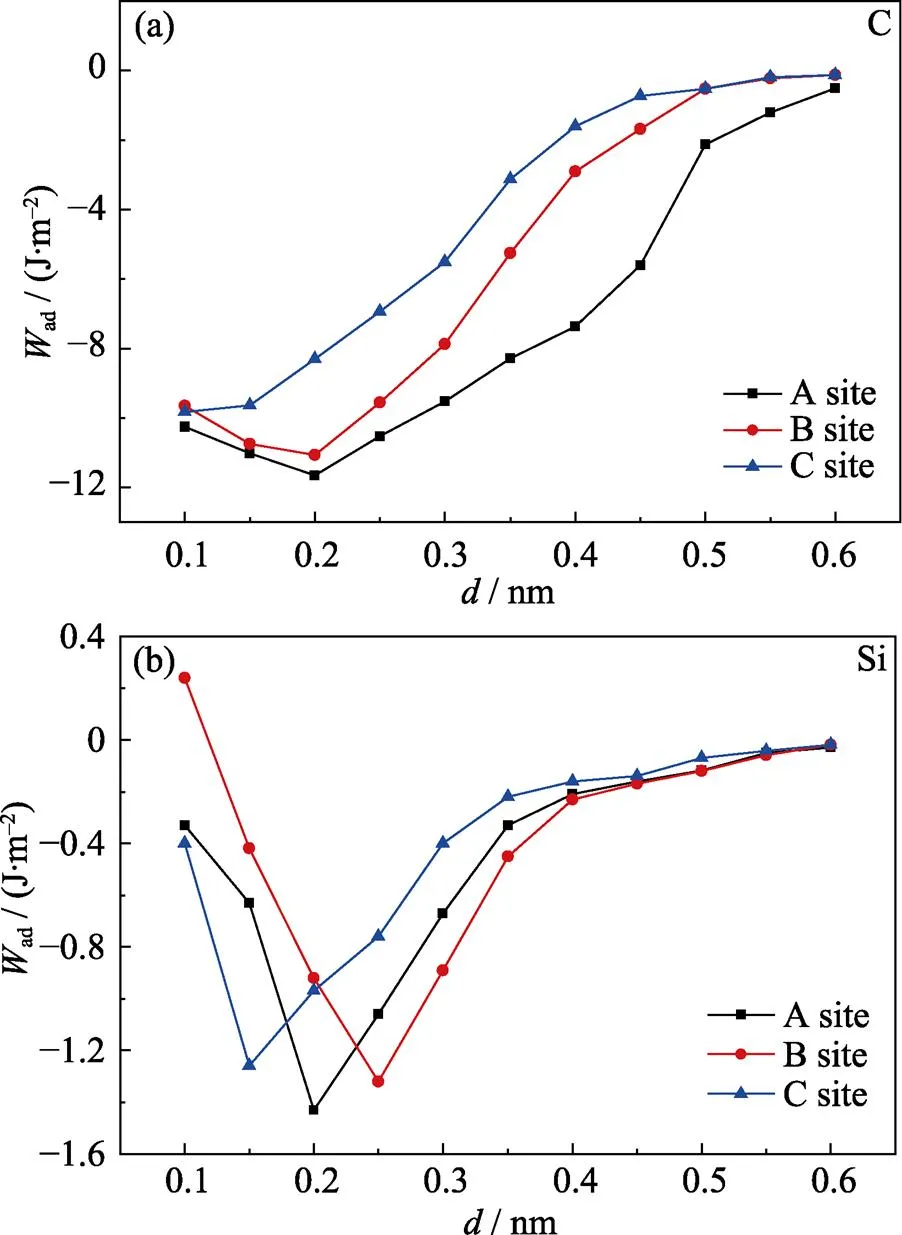
图3 SiC(001)/Al(111)界面分离功与界面距离的关系曲线
Mg、Si、Cu是基体合金常见的添加元素, Saiz等[23]提出的界面吸附驱动润湿理论指出, 合金中活性元素会优先富集在固/液界面处。为了考察合金元素在界面偏聚时对Al/SiC界面结合的影响, 在上述计算优化基础上, 首先构建超晶胞Al/SiC界面模型, 其中以Si原子为终端的bridge-site结构, 界面间距为0.2 nm, 在此优化界面模型上建立三种元素单独掺杂的Al-X/SiC界面结构模型。由于掺杂后越靠近界面的原子越不稳定, 所以需对掺杂后的界面结构再次进行优化, 将建成界面两边的原子均弛豫, 从而达到整个体系原子的充分弛豫及体系能最低。以Mg掺杂为例, 界面处各原子的标号如图4所示。
考虑到取代情况比较复杂, 计算量较大, 本研究只计算了合金原子分别取代界面上一个、两个、四个Al原子。在计算条件完全相同的情况下, 通过比较合金元素取代前后的SiC/Al界面结合能以及电子结构变化, 分析合金元素对SiC/Al复合材料界面结合影响。
3 计算结果及讨论
3.1 Mg、Si、Cu原子掺杂对界面粘着功的影响
表3为Mg、Si、Cu原子掺杂前后SiC/Al体系的粘着功。从表3可知, 当Mg、Si、Cu原子替换界面处Al原子后, 整个体系的能量都会增大。这是因为掺杂Mg、Si、Cu原子后, 晶格将会发生畸变, 产生应力, 导致整个体系的能量增大; 从表3还可以看出随着掺杂原子数目增加, 体系的能量呈线性增大。未掺杂的SiC/Al体系粘着功为851 mJ/m2, 当掺杂一个Mg、Si、Cu原子后, SiC/Al体系粘着功有所增大, 分别为1140 、1024、1088 mJ/m2, 体系粘着功分别提升了34.1%、20.3%、27.8%。这说明Mg、Si、Cu对SiC/Al界面结合均有不同程度的促进作用, 其中掺Mg提升效果最明显, 其次是掺Cu和掺Si; 当掺杂2个Mg、Si、Cu原子时, SiC/Al体系的黏着功达到最大值, 继续增加掺杂个数, 整个体系的黏着功反而有所降低。除Cu掺杂外, 其值均大于未掺杂合金原子体系。随着掺杂原子的增加, 整个体系的黏着功变化规律相同, 均呈先增加后减小的变化趋势。
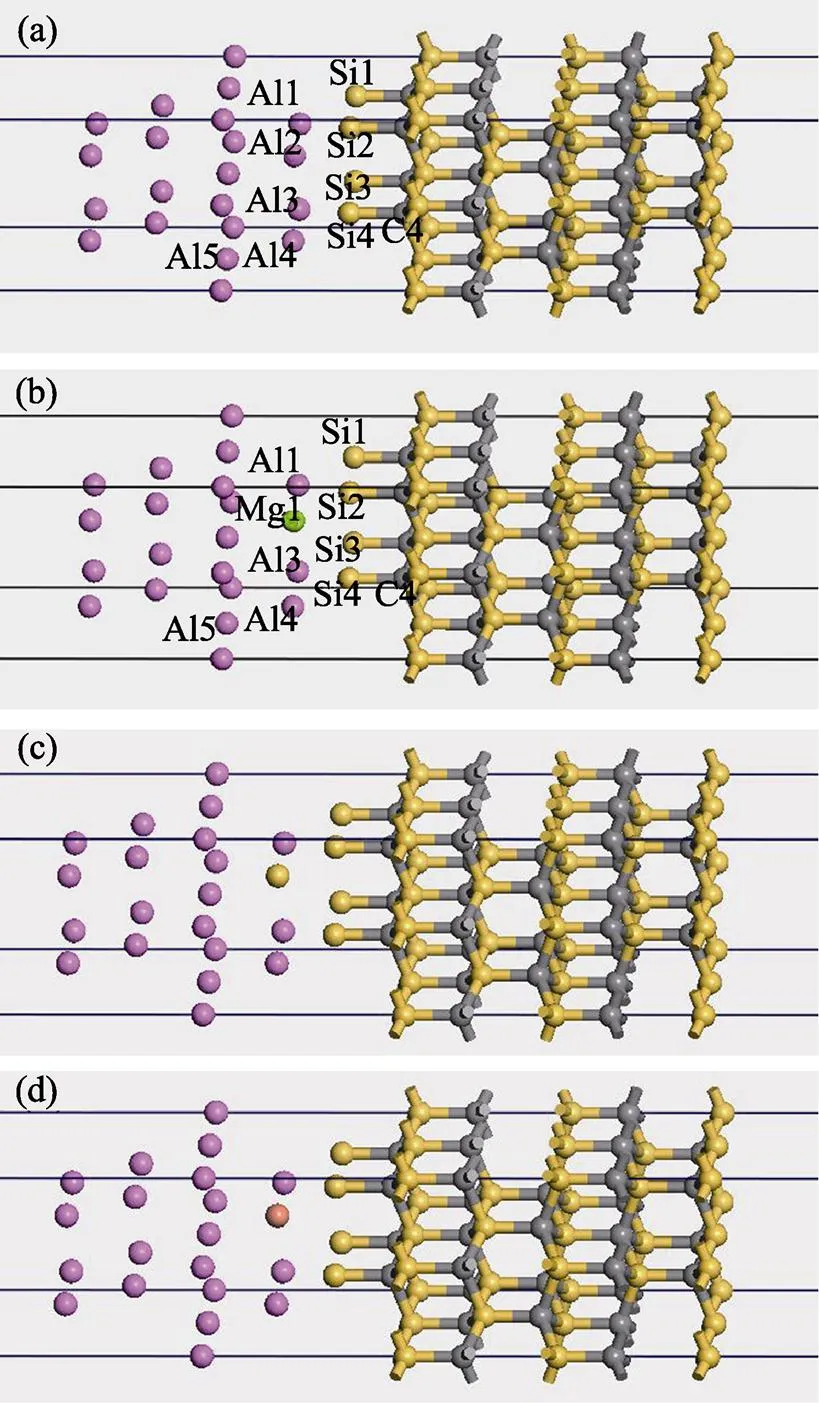
图4 (a)未掺杂、(b)1个、(c)2个、(d)4个Mg原子替换Al原子界面模型
3.2 Mg、Si、Cu原子掺杂对界面处原子的电子结构影响
为了进一步考察Mg、Si、Cu原子对SiC/Al界面结合的影响机理, 本研究分别探讨了掺杂1个Mg、Si、Cu原子的SiC/Al体系的电子结构。以Mg为例, 图5为Mg原子取代界面上Al(2)原子前后, 与此Al原子最近的Al(3)原子以及Si(2)原子的分波态密度PDOS (Density of States)图。
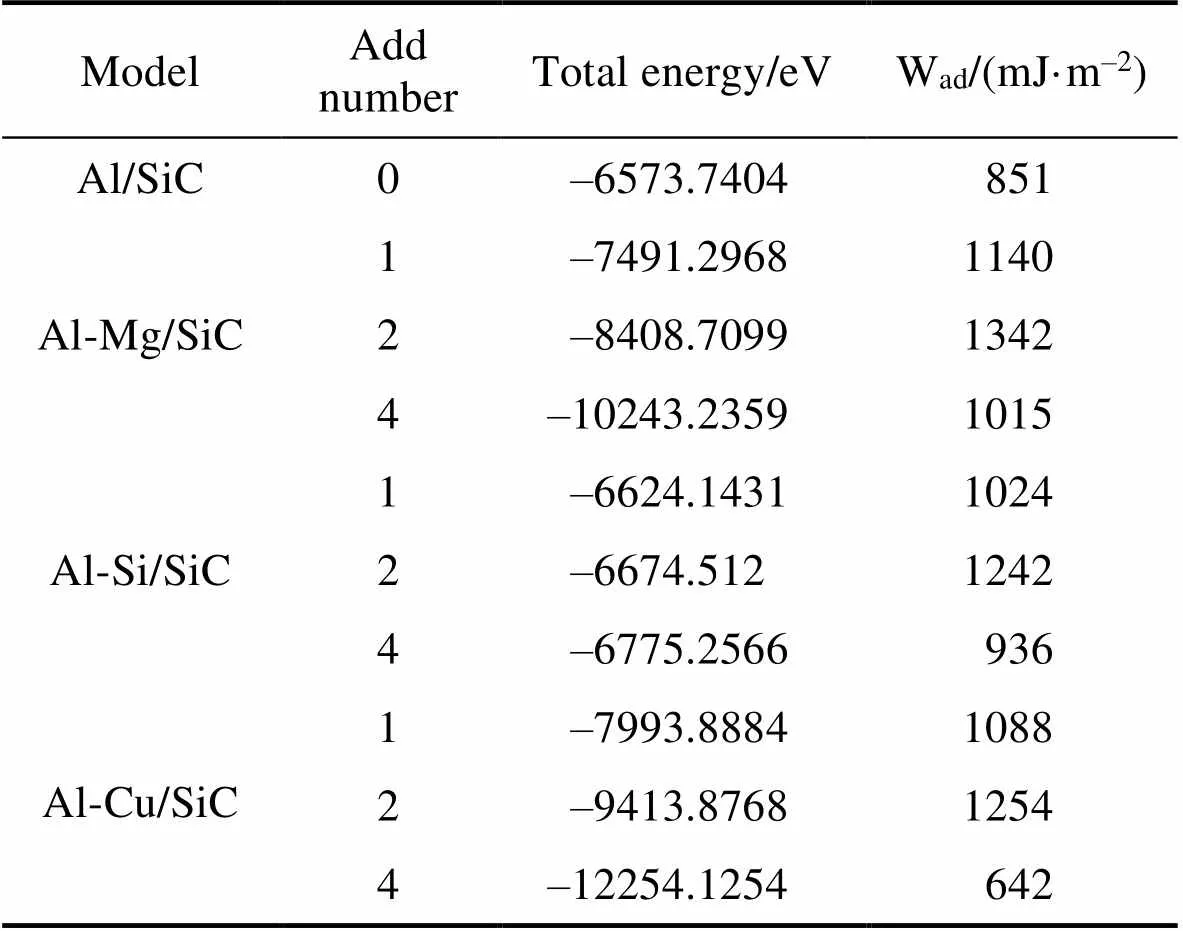
表3 合金原子(Mg、Si、Cu)掺杂前后SiC/Al界面粘着功的变化
从图5(a, b)中可以看出, Mg原子取代Al(2)原子后, 界面上的Al(3)原子s轨道和p轨道电子态密度峰的强度和峰位基本保持不变, 但是峰位向费米能级发生移动, 这表明Al(2)原子中的s电子和p电子能量增加, 活动增强。
与掺杂之前相比, Al原子中 p电子态密度略变宽, 这说明p电子更加自由, 离域性增强, 因此更容易与周围原子成键。在图5(c, d)中, Si(2)原子的s电子和p电子同样向费米能级移动, 同时s轨道费米能级附近的电子态密度明显增加, 这与文献[24]中指出的在界面处因金属原子的诱导, 使得界面的Si原子失去体相的特性, p、s轨道的电荷都增加, 使得Si表现出金属性是一致的。这种状况将有利于Si原子和界面上的Al原子相互作用, 增强界面结合。
表4为合金原子掺杂前后SiC/Al界面处原子的Millken电荷。从表4中可以看出, 当Mg原子替换Al原子后, 界面上Mg原子附近的Al(1)和Al(4)电荷都从2.87 e增加到2.99 e, 且Si(2)、Si(4)的电荷分别从3.98、3.89 e增强到4.04、3.95 e; 当Si原子替换Al原子后, 界面上Si原子附近的Al(1)和Al(4)电荷均从2.87 e分别增加到2.88、2.92 e, 且Si(2)、Si(4)的电荷分别从3.98、3.89 e增强到3.98、3.90 e; 当Cu原子替换Al原子后, 界面上Cu原子附近的Al(1)和Al(4)电荷都从2.87 e分别增加到2.89、2.94 e,且Si(2)、Si(4)的电荷分别从3.98、3.89 e增强到3.99、3.92 e, 这些原子的s、p轨道电荷都有不同程度增加, 因此跨界原子之间相互增强。
单位集居数[25]是表示原子间相互作用的微观参数, 数值越大, 表面原子成键越强。表5为不同合金原子掺杂前后SiC/Al界面及附近成键原子单位集居数。从表5可以看出: 当合金Mg原子掺杂时, 界面处的键Al(1)–Si(1)、Al(3)–Si(3)和Al(4)–Si(4)的单位集居数从0.36分别增加到0.4、0.39和0.4。说明添加Mg原子后, 界面处的Al原子和Si原子之间的相互作用增强, 这与上述电荷计算结果是一致的。
表5中数据还显示界面处的Al(4)原子和亚界面处的Al(5)的以及界面处的Si(4)原子和亚界面处C(4)的集居数分别从0.24和0.24增加到0.25和0.25, 这表明添加Mg原子后, 亚界面处Al和SiC中的原子相互作用也有所增加。
从表5还可以看出, 当合金Si原子掺杂时, 界面处的键Al(1)–Si(1)、Al(3)–Si(3)和Al(4)–Si(4)的单位集居数从0.36分别变化到0.37、0.28和0.34, 这说明添加Si原子后, 界面处的Al原子和Si原子之间的相互作用有所增强, 但与掺杂Mg原子界面处成键的单位集居数相比, 增大幅度有所减小。且与Al-Mg/SiC体系不同的是, 当Si原子替换界面处的Al原子后, 界面处的Al(4)原子和亚界面处的Al(5)的单位集居数反而有所减小, 界面处的Si(4)原子和亚界面处C(4)的单位集居数几乎保持不变, 这表明Si对亚界面层原子和界面层原子的相互作用几乎无影响, 甚至削弱两者之间的结合。当合金Cu原子掺杂时, 同样界面处的Al原子和Si原子之间的相互作用有所增强, 亚界面处Al和SiC中的原子相互作用也所增强。

图5 Mg原子掺杂前后界面处Al和Si原子的PDOS图
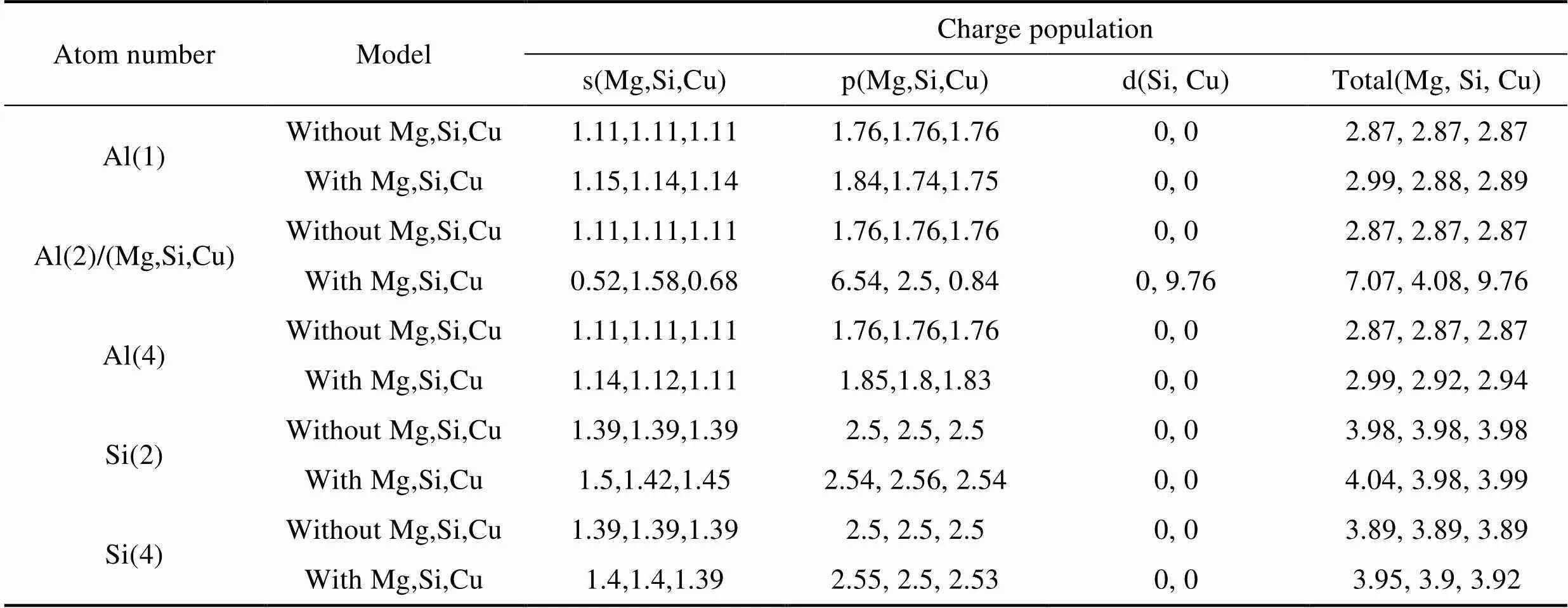
表4 合金原子(Mg、Si、Cu)掺杂前后SiC/Al界面处原子Millken电荷
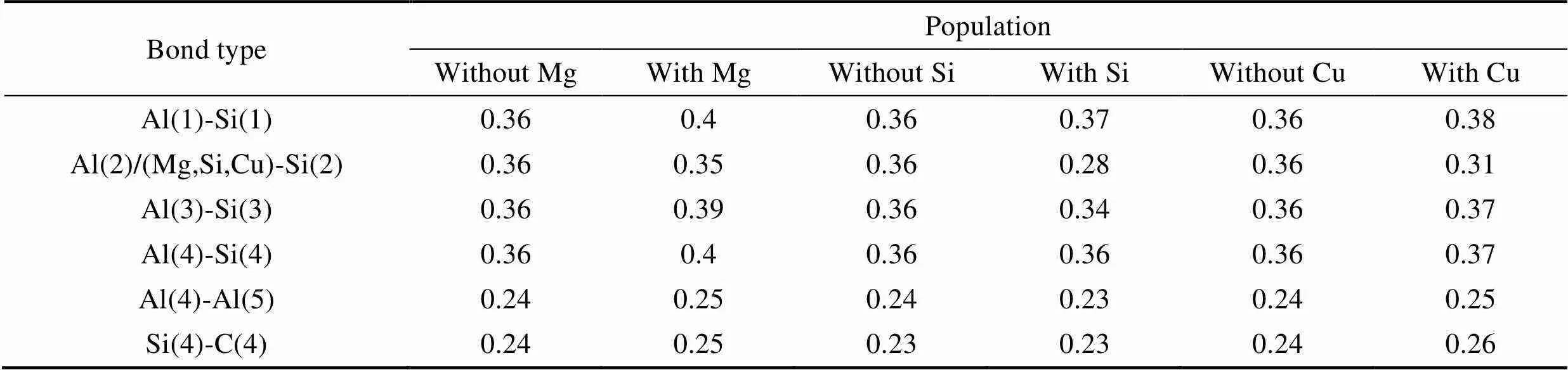
表5 合金原子(Mg、Si、Cu)掺杂前后SiC/Al界面及附近成键原子集居数
3.3 模拟值与实验值的比较
图6为在900 ℃温度下实验测得的纯Al及其添加金属元素合金在-SiC基板上接触角随时间的变化。
从图6中可以看出, 合金元素的添加对Al滴在-SiC基板铺展过程中的初始接触角和最终接触角都有明显的影响。虽然Al-X(X=Mg、Si、Cu)/SiC体系的初始润湿角都大于纯Al/SiC体系的初始润湿角, 不同体系到达平衡状态的时间也不同; 对最终接触角而言, 与纯Al/SiC体系相比, 均有减小, 由原来的73°, 分别变为63°、69°、67°。这进一步佐证基体中添加合金元素(Mg、Si、Cu)能改善Al/SiC体系的润湿性。
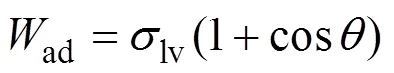
本研究认为添加合金元素后, 模拟值比实验值有偏差的可能原因如下: 由于本实验的测试温度为900 ℃, 在润湿性实验值中, 其计算公式中选取为平衡状态下的接触角, 根据文献[27]报道, 当在平衡状态时, 即已存在一些界面反应, 此时的接触角不仅包含SiC和Al之间的接触角, 而且也包含了中间反应相与Al之间接触角。而本研究中第一性原理计算时还未包含界面新相的影响, 所以存在一定的偏差。
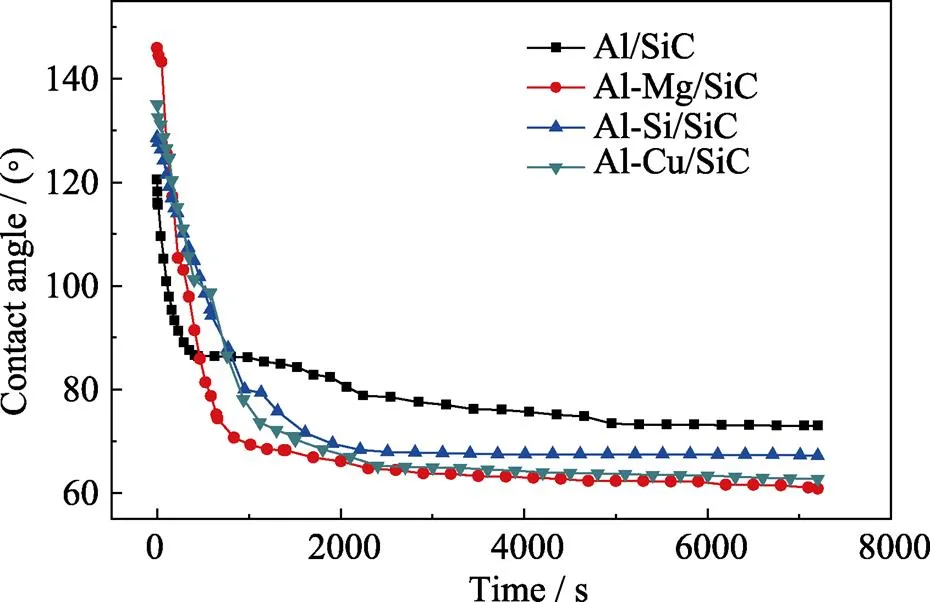
图6 900 ℃时纯Al及其合金(Al-Mg、Si、Cu)在α-SiC基板上接触角随时间的变化

表6 不同体系界面黏着功
4 结论
1) Al(111)面和SiC(001)面是各自低指数面中表面能最小的晶面。在SiC(001)面, 以Si原子为终止面的表面能小于以C原子为终止面的表面能, 其界面粘着功则总体比以 C原子为终止面的小; 当以Si原子为终止面的Brigde结构时, 其界面粘着功与实验值相吻合是SiC/Al结合的主要方式。
2) Mg、Si、Cu三种元素的掺杂使得界面黏着功增加, 界面处原子之间的单位布居数增大, 互相成键作用加强, 使得SiC/Al体系更加稳定。掺Mg后体系黏着功最大, 其次是掺Cu和掺Si。随着掺杂合金原子数量增加, SiC/Al体系的黏着功都呈先增大后减小的变化趋势。
3) Mg、Si、Cu三种元素的添加虽然会使SiC/Al体系初始接触角有所增加, 但最终接触角均小于纯Al/SiC体系。最终接触角由73°, 分别变为63°、69°和67°; 第一性原理计算的合金元素掺杂前后, 体系的黏着功和实验值较为接近, 且变化规律相同。
[1] SONG M, HE Y H. Effects of die-pressing pressure andextrusion on the microstructures and mechanical properties of SiC reinforced pure aluminium composites., 2010, 31(2): 985–989.
[2] FOX R T, NEWMAN R A, PYZIK A J,. Al2O3-B4C-Al composite material systempressure less infiltration methods.., 2010, 7(6): 837–845.
[3] LEE H S, JEON K Y, KIM H Y,. Fabrication process and thermal properties of SiCp/Al metal matrix composites for electronic packaging applications., 2000, 35(24): 6231– 6236.
[4] LI C Y, HU Y K, ZHENG X J,. Study of microstructure of SiCp/ZL101A composites by vacuum hot-pressing sintering processing.,2014, 41(2): 413–416.
[5] HE P, HUANG S Y, WANG H C,. Electroless nickel –phosphorus plating on silicon carbide particles for metal matrix composites.,2014, 40(10): 16653–16664.
[6] BHUSHAN R K, KUMAR S, DAS S. Optimisation of porosity of 7075 Al alloy 10% SiC composite produced by stir casting process through Taguchi method., 2009, 1(1): 116–129.
[7] MANDALl D, VISWANATHAN S. Effect of re-melting on particles distribution and interface formation in SiC reinforced 2124Al matric composite.,2013, 86: 21–27.
[8] RATNAPARKHI P L, HOWE J M. Characterization of a diffusion- bonded Al-Mg alloy/SiC interface by high resolution and analytical electron microscopy., 1994, 25(3): 617– 627.
[9] LUO Z P. Crystallography of SiC/MgAl2O4/Al interfaces in a pre-oxidizied SiC reinforced SiC/Al composite., 2006, 54(1): 47–58.
[10] MANDALl D, VISWANATHAN S. Effect of heat treatment onmicrostructure and interface of SiC particle reinforced 2124 Al matrix composite., 2013, 85: 73–81.
[11] AGUILAR-MARTÍNEZ J A, PECH-CANUL M I, RODRÍGUEZ- REYES M,. Effect of Mg and SiC type on the processing of two-layer Al/SiCp composites by pressureless infiltration., 2004, 39(3): 1025–1028.
[12] EUSTATHOPOULOS N, LAURENT V, RADO C. Wetting kinetics and bonding of Al and Al alloys on-SiC.,1996, 205(1/2): 1–8.
[13] KOBASHI M, CHOH T. The wettability and the reaction for SiC particlce/Al alloy system.,1993, 28(3): 684–690.
[14] FERRO A C, DERBY B. Wetting behavior in the Al-Si/SiC system: interface reactions and solubility effects.., 1995, 43(8): 3061–3073.
[15] MA X C, WU J B. An investigation on wettability and interfacial phenomena of Al-SiC system., 1994, 12(1): 37–41.
[16] ZHANG Q, JIA L T, WU G H. Fabrication of oxidized SiC particles reinforced aluminum matrix composite by pressureless infiltration technique.., 2012, 27(4): 353–357.
[17] LIU J Y, LIU Y C, LIU G Q,. Oxidation behavior of silicon carbide particales and their interfacial characterization in aluminum matrix composites.., 2002, 12(5): 961–966.
[18] ZHANG D, SHEN P, SHI L X. Wetting and evaporation behaviors of molten Mg on partially oxidized SiC substrates., 2010, 256(23): 7043–7047.
[19] JONES R O, GUNNARSSON O. Density-functional formalism: sources of error in local-density applications., 1989, 61(3): 689–746.
[20] HONG T, SIMTH J R, SROLOVITZ D J. Theory of meta-ceramic adhesion.,1995, 43(7): 2721–2730.
[21] HAYES R L, ORTIZ M, CARTER E A. Universal binding-energy relation for crystals that accounts for surface relaxation.,2004, 69(17): 1324–1332.
[22] SHI L X, SHEN P, ZHANG D, JIANG Q C. Wetting and evaporation behaviors of molten Mg-Al alloy drops on partially oxidized-SiC substrates., 2011, 130(3): 1125–1133.
[23] SAIZ E, CANNON R M, TOMSIA A P. Reactive spreading in ceramic/metal systems..,2001, 56(1): 89–96.
[24] HOEKSTRA J, KOHYAMA M.calculations of the-SiC(001)/Al interface., 1998, 57(4): 2334–2341.
[25] WINKLER B, PICKARD C J, SEGALL M D,. Density- functional study of charge disordering in Cs2Au(I)Au(Ⅲ)Cl6under pressure., 2001, 63(21): 214103–214106.
[26] LIU Y M, SHI J Y, LU Q Q,. Research progress of solid surface energy calculation based on Young's equation.., 2013, 23(11): 123–129.
[27] CANDAN E, ATKINSON H V, TUREN Y,. Wettability of aluminum-magnesium alloys on silicon carbide substrates..,2011, 94(3): 867–874.
Alloy Elements on SiC/Al Interface: a First-principle and Experimental Study
ZOU Ai-Hua, ZHOU Xian-Liang, KANG Zhi-Bing, RAO You-Hai, WU Kai-Yang
(School of Material Science and Engineering, Nanchang Hangkong University, Nanchang 330063, China)
First-principle approach based on density functional theory and experimental method were used to study the interface bonding of SiC/Al substituted by alloy elements Mg, Si and Cu. The electronic structure and bonding of alloy elements at interface segregation were investigated. The results show that bridge-site model with bridge Si is the most stable combination way of SiC/Al interface after optimization of the interface structure of pure Al/SiC system. When Al atoms at the interface replaced by alloy elements separately, the electronic structural parameters such as partial density of states, Mulliken charge and bonding population, all vary in different degree, which not only increase the binding of Si and Al atoms at the interface, but also increase the interaction among alloy atoms, Al matrix and SiC reinforcement at the interface and sub-interface. It is conducive to enable the system more stable and the adhesion work of interface more differently. Among them, the increased adhesion work is the most obvious when Mg is doped, followed by Cu and Si. Furthermore, the adhesion work of Al/SiC systems doped alloy elements calculated by first-principle is close to the experimental values, and the law of change is the same.
alloy element; SiC/Al interface; interface bonding; first principle
TB333
A
1000-324X(2019)11-1167-08
10.15541/jim20180540
2018-11-12;
2019-03-08
国家自然科学基金(51562027); 江西省自然科学基金(20171BAB216003); 江西省教育厅基金(GJJ160685)National Natural Science Foundation of China (51562027); Natural Science Foundation of Jiangxi Province (20171BAB216003); Scientific Research Project of the Education Department of Jiangxi Province (GJJ160685)
邹爱华(1980–), 女, 博士, 副教授. E-mail: aihua553030@163.com
猜你喜欢
石材(2022年3期)2022-06-01 06:23:54
原道(2022年2期)2022-02-17 00:59:12
原子与分子物理学报(2021年1期)2021-03-29 07:28:30
煤炭学报(2021年2期)2021-03-24 02:22:32
理化检验-化学分册(2020年5期)2020-06-15 11:36:04
电镀与环保(2018年4期)2018-08-20 03:08:02
实用口腔医学杂志(2017年6期)2017-09-19 02:51:14
中华老年口腔医学杂志(2016年4期)2017-01-15 14:25:13
西南石油大学学报(自然科学版)(2016年6期)2017-01-15 14:14:19
中国石油大学学报(自然科学版)(2015年2期)2015-11-10 06:08:25
