
CRISPR-Cas生物传感器研究进展
2019-12-11 06:12李凯罗云波许文涛
生物技术进展 2019年6期
李凯,罗云波,许文涛*
1.中国农业大学食品科学与营养工程学院,北京食品营养与人类健康高精尖创新中心,北京 100083;2.中国农业大学,农业农村部农业转基因生物安全评价(食用)重点实验室,北京 100083
CRISPR系统源于细菌体内的免疫系统,能够对外来核酸分子进行识别和剪切。由于CRISPR系统特异性的识别机制,研究人员将其开发成为新型的基因编辑技术,使其成为最热的分子生物学工具。以CRISPR-Cas9体系为代表的编辑系统已经成功应用于农业、医学、生物学等领域,如用于建立新型育种方式、研制新型基因药物以及探索生物体内通路等。CRISPR应用最多的为基因编辑领域,CRISPR技术能够准确的靶向目标实现定点切割,促使胞内自动修复系统完成对靶标序列碱基的替换和缺失。近年来,虽然CRISPR技术脱靶效应产生的潜在风险逐渐被发现,但是随着该技术的不断发展和完善,问题也会随之逐步解决。CRISPR与靶标序列的特异性结合方式成为了一种特殊的信号识别机制,可以应用在生物传感领域。越来越多的研究人员开始以CRISPR系统为基础,与其他技术结合在一起,如等温扩增技术(recombinase polymerase amplification)、电化学传感、光学显色等,以建立特殊的生物传感器。此类传感器具有识别特异性强、检测灵敏度高、简单方便等特点。因此本文综述了基于CRISPR-Cas生物传感器的研究进展,以期为生物传感器的相关研究提供参考。
1 CRISPR-Cas系统
1.1 CRISPR-Cas系统的发现
CRISPR/Cas是来源于一类广泛存在于细菌及古细菌中的适应性免疫系统。1987年,日本科学家Ishino等[1]克隆并测序了iap基因,该基因负责大肠杆菌中碱性磷酸酶同工酶的转化。随后他们在研究中克隆并测序发现,在iap的下游有一组由29个核苷酸组成的重复序列被非重复、类似的短序列分开,这是首次对CRISPR特殊结构进行报道。在2002年,Jansen等[2]创造了术语“CRISPR”以反映这些特定结构。通常,重复簇之前是“前导”序列,AT富集区域有几百个碱基对,具有种内保守性,但不具有种间保守性。CRISPR保守蛋白编码基因,通常存在于整个基因座的一侧。对几个CRISPR基因座中间隔序列的分析表明,间隔区匹配来自外源遗传元件的序列,如噬菌体和质粒[3]。2007年,Barrangou等[4]的一项开创性研究首次证明了CRISPR序列是一种适应性免疫途径,可保护嗜热链球菌免受噬菌体感染。Cas位点编码的多个核酸酶和解旋酶可以把入侵的噬菌体DNA 切割,然后整合到CRISPR 的重复序列中。当下次再遇到相同噬菌体入侵时,细菌转录RNA-Cas 蛋白复合物利用这些与入侵噬菌体DNA 同源的RNA去切割外源的DNA,从而达到识别并对抗噬菌体感染的目的。
1.2 CRISPR-Cas系统分类
CRISPR-Cas系统包含多种类型,CRISPR-Cas系统大致分为两大类(图1):1类系统依赖于多亚基蛋白质复合物共同作用,而2类系统利用单个蛋白质。在这两类系统中,根据能够表达Cas蛋白的种类的不同,到目前为止可分为I型、II型、III型、IV型、V型和VI型(图1未包含VI型),其中I、III、IV属于第一大类,II、V、VI属于第二大类。随着对细菌免疫系统基因组功能的不断挖掘,新型的CRISPR-Cas系统也逐渐被认识。
I型CRISPR-Cas系统中具有特殊的cas3基因,能够编码出同属具有解旋酶活性以及DNase活性的大分子蛋白质。I类系统中还具有编码多种其他蛋白的基因包括cas5、cas6和cas7,这些基因表达出来的蛋白会与Cas3蛋白相互结合形成复杂复合体共同行使作用。此外,参与CRISPR-Cas功能的复合蛋白中可能还包括BH0338,这类大蛋白家族以及Cse1和Cse2这类小的α-螺旋蛋白。CRISPR-Cas系统中表达的串联蛋白复合体能够加工前体crRNA,使之转化为成熟crRNA。
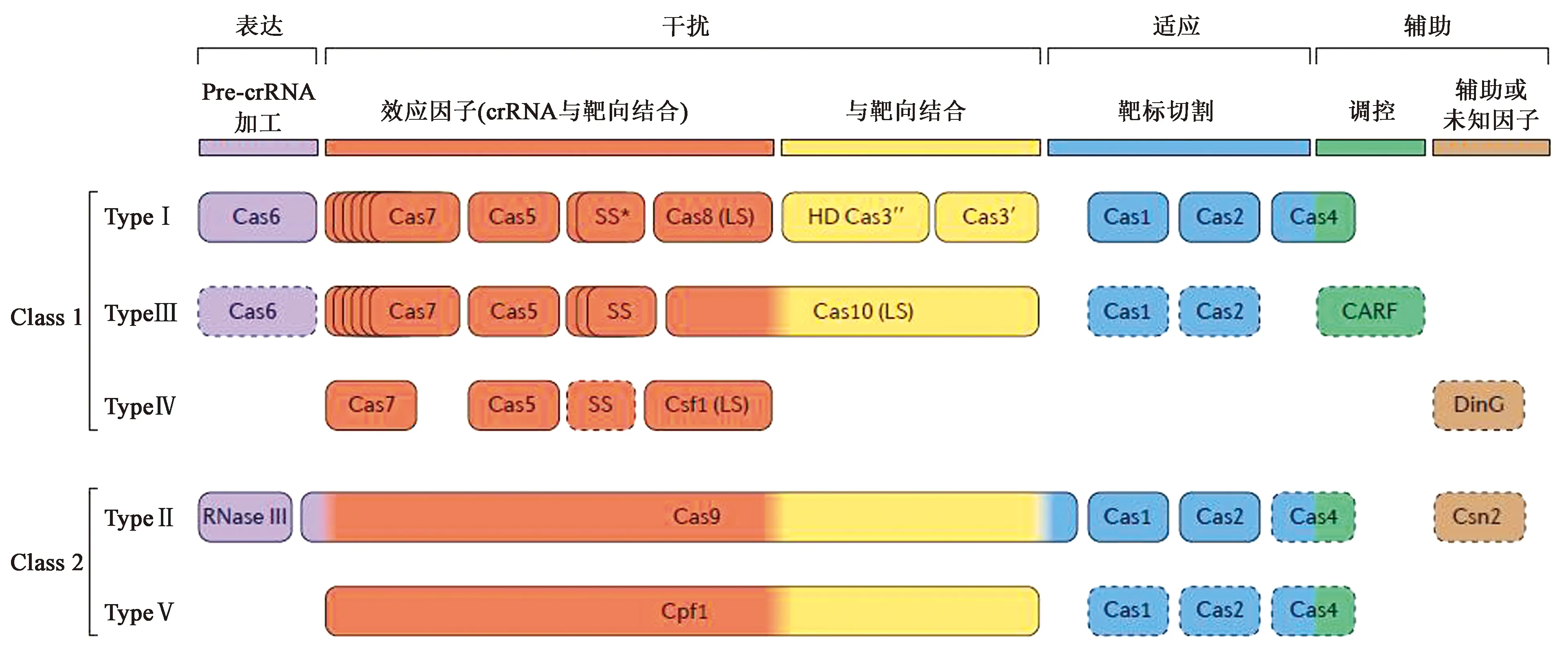
图1 CRISPR-Cas系统分类图[5]Fig.1 Classification figure of CRISPR-Cas system[5]
II型CRISPR-Cas系统中最为人熟知的是CRISPR-Cas9。II类系统中包含特殊的cas9基因以及csn基因,根据csn的不同类型可将II类系统分为两个亚型,分别包含csn2和csn4基因。Cas9蛋白是一种功能十分强大的蛋白质,具有识别crRNA以及切割靶标DNA的能力。Cas9蛋白具有两个核酸内切酶的结构域,位于蛋白N端的RuvC和中部NHN,分别负责切割外源DNA与间隔序列的互补链和外源DNA的另外一条链。在整个切割过程中,首先tracrRNAs和前体crRNA中的重复序列杂交,再与Cas9蛋白进行结合,通过RNaseIII对RNA复合体进行切割,形成不同类型的成熟crRNA-tracrRNA-Cas9复合体。形成的成熟复合体再通过PAM序列识别进行外源DNA切割。
III型CRISPR-Cas系统以cas10基因为标志基因,先前根据可以表达csm2和cmr5两种亚基分为两个亚型:III-A和III-B。现已证明亚型III-A和亚型III-B CRISPR-Cas系统可以靶向RNA和DNA。后来研究人员发现了另外两种III型系统:III-C和III-D。III-C亚型的显著特征是Cas10蛋白的环化酶活结构域的明显失活。亚型III-D基因座通常编码缺乏HD结构域的Cas10蛋白,但它们还含有一种独特的cas5基因,也称为csx10。这两种新亚型都缺少cas1和cas2基因,但整个系统具有完整的功能性。
IV型CRISPR-Cas系统是在不常见的几种细菌的质粒上发现的一种新型CRISPR-Cas系统,csf1基因是该系统的标志性基因。系统基因座的组成大部分与III-B亚型类似,缺少cas1和cas2基因。IV型系统可以编码出最小的蛋白复合体对crRNA进行加工,其中包括Csf1、Cas5和Cas7蛋白。IV型CRISPR-Cas系统有两种不同的亚型,其中一种含有DinG家族解旋酶,第二种缺乏DinG但通常含有编码小α-螺旋蛋白的基因,类似于亚型III-B系统,可以对多种重复回文系列进行crRNA的加工。
V型CRISPR-Cas系统是基于在一种古细菌基因组发现的独特cpf1基因,后续也在几种不同细菌基因组中发现了该系统的基因序列。该系统中cpf1基因与cas1、cas2、cas4和CRISPR序列相邻,能够表达Cpf1蛋白。Cpf1包含约1 300个氨基酸,具有跟Cas9相类似的RuvC结构域,并且与精氨酸富集结构和锌指结构相连接。虽然Cpf1缺乏Cas9蛋白中的HNH核酸酶结构域,但V型系统能够对外源DNA进行准确的识别和切割,从而对细菌起到保护防御的作用[6]。
VI型CRISPR-Cas系统是2015年张锋与俄国科学家Koonin通过电脑分析得出的一种新型的CRISPR-Cas系统[7],包括cas1、cas2、c2c2和CRISPR序列。该系统的特殊基因为c2c2基因,能够编码C2C2蛋白。该蛋白具有两个HEPN(higher eukaryotes and prokaryotes nucleotide-binding)结构域,具有RNase活性能够实现对RNA的识别和剪切。通常认为该系统是在细菌防御外源核酸入侵时,针对RNA设立的第二防线。
1.3 CRISPR-Cas生物传感器在检测成像中的应用前景
核酸作为重要的生物标志物,其在体内的含量信息和位置信息都是相关疾病诊断中重要的参考条件。针对于DNA或者RNA检测成像技术的开发,已经成为重要的研究领域之一。以往的检测成像技术以体外扩增、蛋白杂交、微矩阵等技术为基础,虽然可以实现对于靶标的检测成像要求,但是在时间、成本、精准度、灵敏性方面都存在着一定的缺陷,所以需要建立更加快速准确的检测成像新手段。
随着CRISPR-Cas在细菌免疫过程中的相关机制研究越来越清晰,以及多种类型的Cas蛋白不断发现,CRISPR-Cas复合体对于外源DNA和RNA的高效特异性识别机制引起了研究者们的关注。例如双链DNA序列特异性的Cas9蛋白酶、Cas12a蛋白酶,以及RNA序列特异性的Cas13a蛋白酶等。通过对CRISPR的体外定向修饰,建立了一定的逻辑机制,再结合不同剪切属性的Cas酶可以建立起一系列基于CRISPR-Cas技术的检测和成像传感器,此类传感器利用CRISPR-Cas高特异性的识别机制,能够满足高精准检测成像要求,在疾病预防诊断方面具有广阔的应用前景。
2 CRISPR-Cas9生物传感器介导的检测技术
CRISPR-Cas9系统由于Cas9蛋白的多功能特性,利用其DNA靶向剪切产物进行循环放大以及信号输出的方式可以对DNA核酸进行高效检测。东南大学生物电子学国家重点实验室王进科教授团队利用Cas9蛋白的这一性质,建立了CRISPR-PCR检测方法靶向检测人乳头瘤病毒DNA[8]。在人乳头瘤病毒保守序列上选取两处sgRNA (single guide RNA)识别区域,建立sgRNA-Cas9复合物。首先利用PCR技术将该保守序列进行初步扩增,将PCR产物中加入sgRNA-Cas9复合物进行特异性切割,剪切产物利用Taq酶在末端链接鸟苷酸,并将剪切产物连接到T载体上,最后利用含有T载体以及切割产物序列的特异性引物对连接好的质粒进行PCR,最终实现5 ng靶标DNA的检测(图2)。该方法还同样适用于qPCR进行实时荧光检测。
与上述方法原理类似,王进科教授团队还建立了一种基于CRISPR-Cas9技术的反向PCR技术应用于DNA检测[9]。该方法在靶标DNA片段上游、下游分别设计一条sgRNA,当Cas9蛋白发生特异性剪切后会形成双链片段,利用双链片段设计的反向引物进行PCR扩增可以使剪切产品进行分子内成环或形成多片段产物(实验验证产物为环状结构),在通过电泳或者qPCR荧光进行结果的信号输出(图3)。该方法可以在1.5 h内检测到0.02 ng的样品。
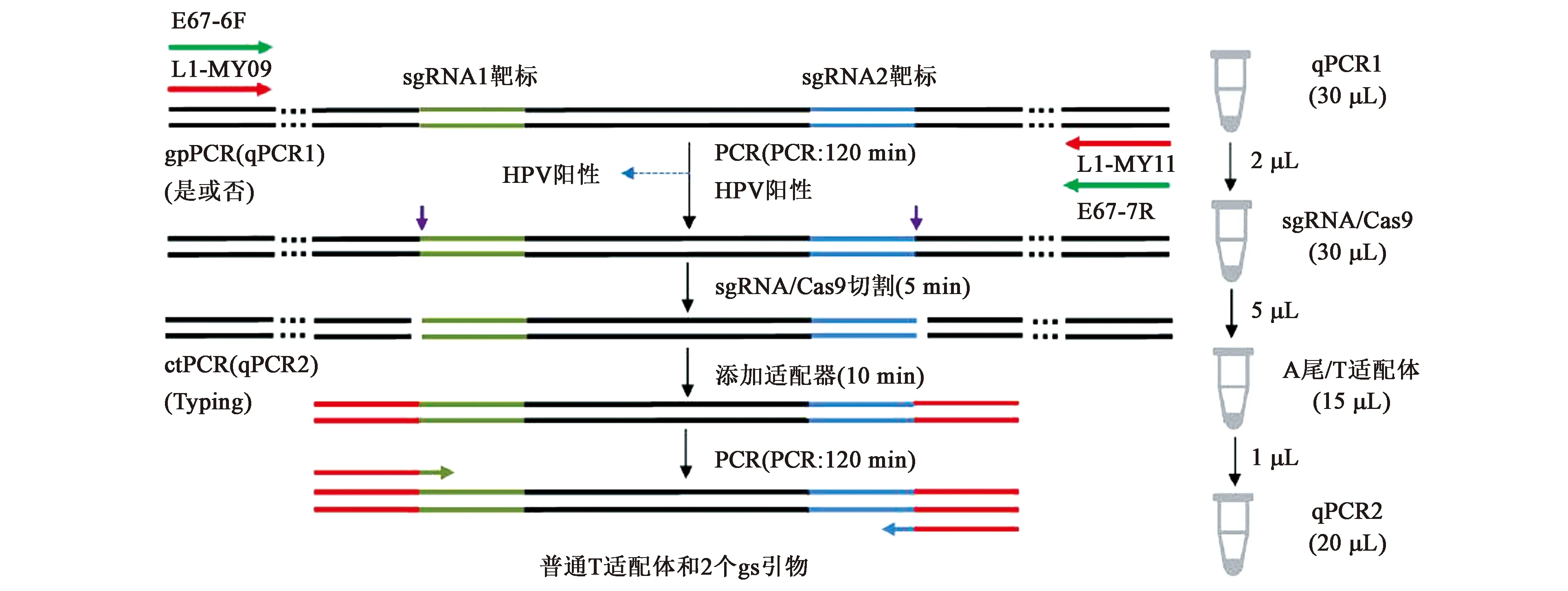
图2 CRISPR-PCR检测原理图[8]Fig.2 The detection principle of CRISPR-PCR[8]
由于CRISPR-Cas9在识别基因组低拷贝靶标基因时,其识别以及剪切的效率较低,为将靶标含量提高到CRISPR-Cas9有效范围内,往往会在进行Cas9剪切之前利用PCR或等温扩增技术进行靶标的初步扩增,CRISPR-Cas9系统仅用于终点分析,这在很大程度上限制了检测技术的发展。鉴于此,中国科学院深圳先进技术研究院生物医学材料与界面中心喻学锋研究员团队,建立了CRISPR-Cas9触发的切口核酸内切酶介导的SDA(strand displacement amplification)方法(CRISDA),用于灵敏扩增和检测双链DNA[10]。CRISDA技术充分利用了具有高灵敏度、高特异性和高独特性的CRISPR-Cas系统识别靶DNA并释放单链结构的特性,结合稳健的肽核酸(PNA)介导的终点测量,在复杂样品背景下实现了amol级检测和单碱基检测。在进行了一系列概念验证后,CRISDA能够在人体基因组、转基因玉米基因组等复杂背景条件下实现靶向的超灵敏检测。首先,设计一对sgRNA(sgUPS/DNS)与Cas9蛋白结合,识别靶DNA的每个边界并在两个非靶标链中诱导一对切口(图4,步骤1)。随后,引入一对引物以启动SDA反应(起始引物对,IPUPS/DNS)并与暴露的非靶标链杂交(步骤2)。每个IP引物含有5′Nb.BbvCI核酸内切酶切口位点,与暴露的非靶链互补的中间杂交区和与非靶链的双链区互补的3′突出端。加入SDA混合物后,DNA聚合酶在单链DNA结合蛋白TP32的帮助下沿非靶链延伸IP引物。同时,暴露的非靶链也通过DNA聚合酶从Cas9诱导的切口位点延伸到退火的IP引物的5′末端,产生双链Nb.BbvCI核酸内切酶切口位点。另外,Nb.BbvC、聚合酶和SSB一起反Cas9结合位作用以沿着靶DNA给予相点的线性链置换反应(步骤3)。然后,将线性置换的单链Strand-For和Strand-Rev分别与引物IPDNS和IPUPS退火,进一步在90 min内诱导所选靶序列的指数扩增,得到产物1~3(步骤4)。最后,添加靶向扩增子的生物素标记和Cy5标记的PNA探针,并在简单的磁性下拉后通过Cy5的荧光强度定量测定扩增的靶标DNA(步骤5)。
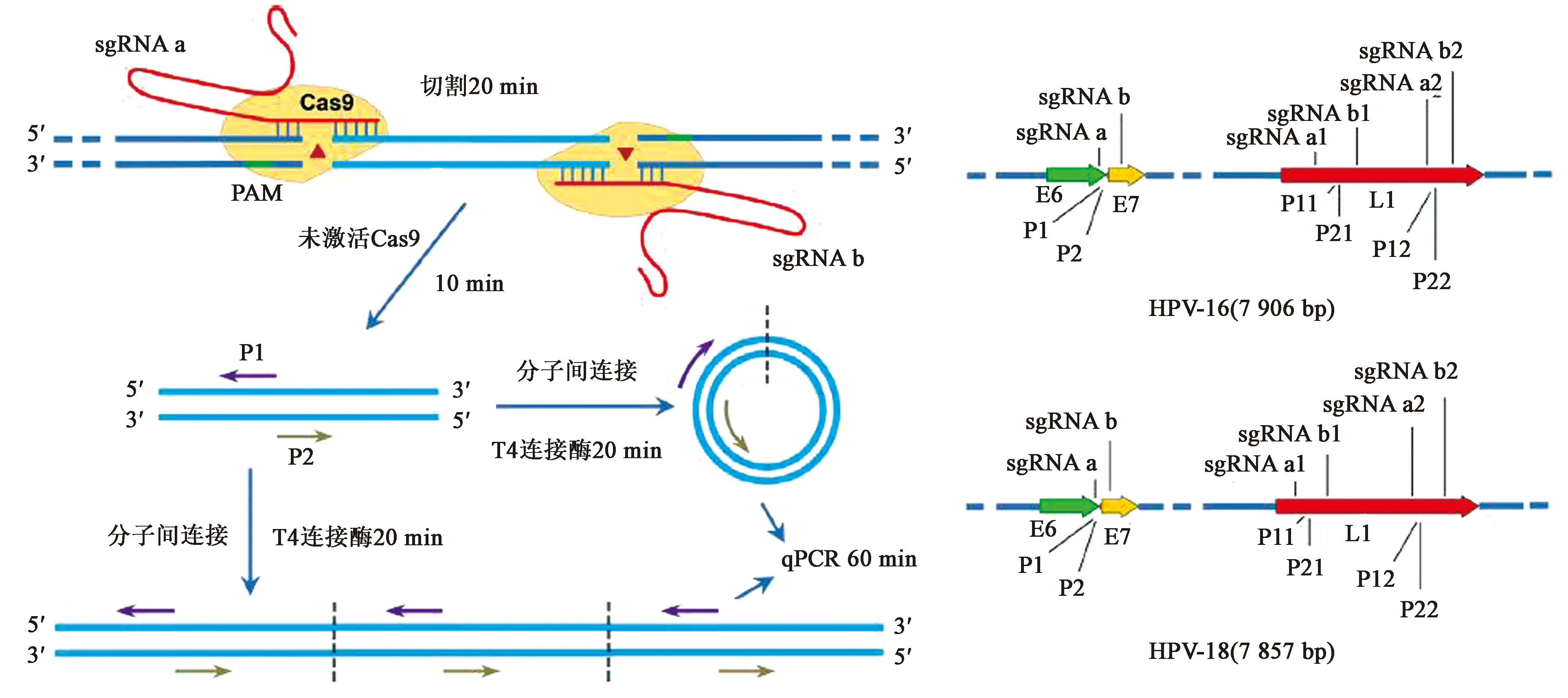
图3 CRISPR介导的反向PCR检测原理图[9]Fig.3 The principle of reverse PCR detection based on CRISPR[9]
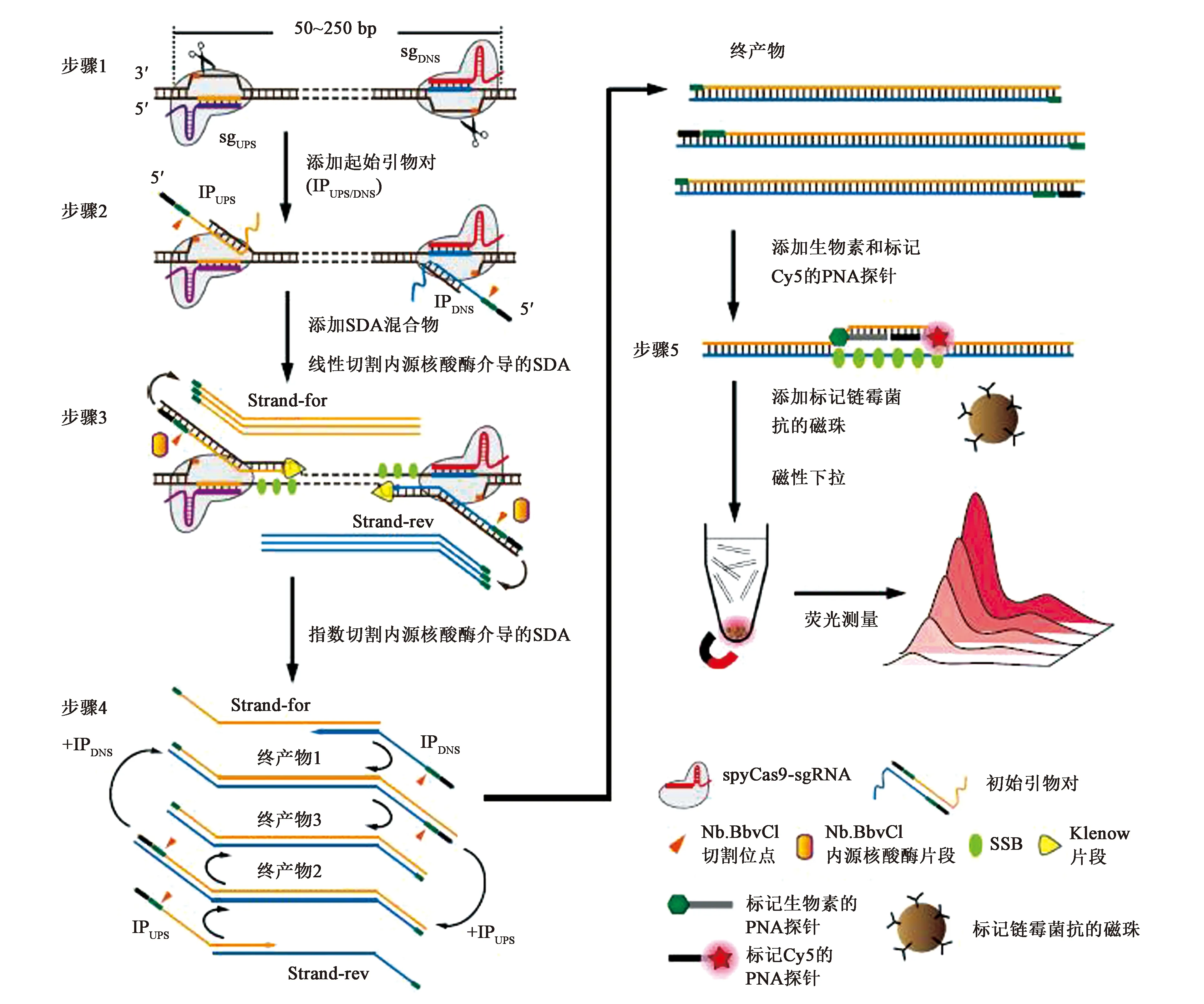
图4 CRISDA超灵敏检测DNA原理图[10]Fig.4 The principle of ultrasensitive DNA detection based on CRISDA[10]
CRISPR-Cas9系统与等温扩增技术相结合可以有效提高检测灵敏度,实现超灵敏检测。但是,在实际基因组DNA检测中,PAM位置的不可选择性为建立CRISPR-Cas9切割位点制造了障碍。不过有报道称,Cas9-sgRNA复合物在有PAM独立单链存在的条件下,就可以对ssDNA或者ssRNA进行特异性识别和剪切[11-12]。根据这个特点,华南师范大学邢达教授团队在利用人为添加PAM序列的基础上,将CRISPR-Cas9系统与恒温指数扩增(isothermal exponential amplification reaction,EXPAR)相结合建立了CAS-EXPAR核酸检测模型[13]。整个反应原理如图5A,使用与PAM序列互补的李斯特菌溶血素基因片段作为靶模型。首先,sgRNA被设计为包含三个区段:20 nt指引序列、30 nt双链互补区域和3个tracrRNA茎环。sgRNA可以与Cas9结合以形成活化的Cas9/sgRNA复合物,其可以进行靶标的位点特异性切割。其次,设计EXPAR模板(T)以执行随后的指数扩增,其中心位置具有NEase识别序列,两段式能够与Cas9切割的靶片段(X)互补的侧翼区(X′)。通过这些设计,X与T的3′末端区域的X′杂交形成复合物(TX),并作为DNA聚合酶的引物,合成具有完整识别序列的双链DNA。然后,NEase在形成的dsDNA中切割T的互补链,并且DNA聚合酶可以依次延伸切割的dsDNA,同时置换X。释放的X可以与另一个T杂交以再次触发扩增。通过CRISPR/Cas9介导的切割、NEase切割、聚合酶延伸和链置换的反应,可以产生大量的dsDNA。结果表明,该方法的检出限为0.82 amol,并且在鉴别单碱基错配方面表现出极佳的特异性。利用相同原理,华南农业大学刘英菊教授团队建立了基于CRISPR-Cas9技术的比色传感器并成功用于鉴定致病疫霉基因组DNA[14]。该方法利用CRISPR-Cas9特异性切割出发扩增反应并将金纳米颗粒(AuNPs)作为光学探针,最后通过AuNPs的聚散程度导致颜色从酒红色到紫色的变化进行判断(图5B)。可视化检测极限为2 pmol,通过吸光度测量值判断的线性范围在0.2 pmol/L~20 nmol/L之间。
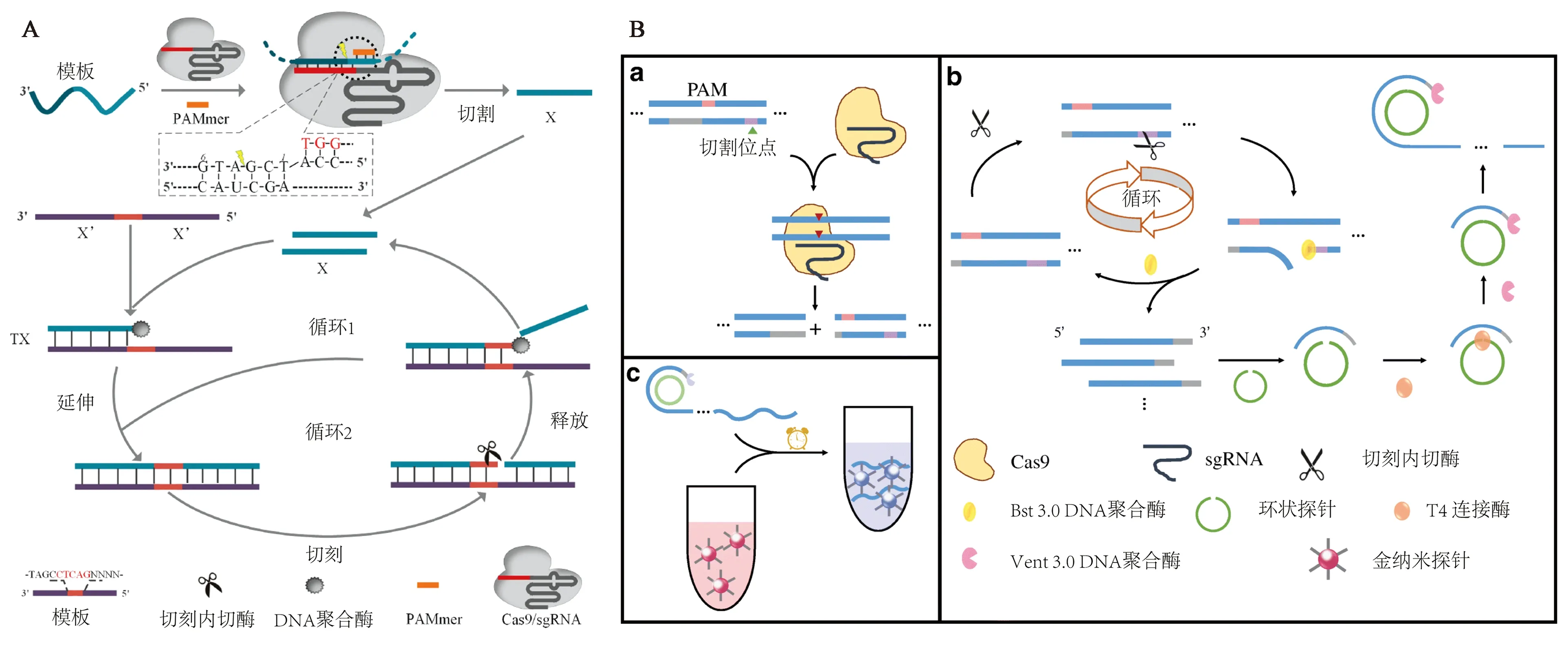
A:CRISPR-EXPAR反应原理图;B:CRISPR识别机制偶联RCA滚环扩增实现核酸显色检测 a—CRISPR-Cas识别、剪切特异性位点;b—EXAPR实现单链富集并触发RCA滚环扩增;c—金纳米富集产生颜色变化。图5 CRISPR-EXPAR反应原理与CRISPR识别机制偶联RCA滚环扩增实现核酸显色检测[13-14]Fig.5 The principle of CRISPR-EXPAR and color method of DNA detection based on CRISPR-EXPAR assay[13-14]
3 CRISPR-dCas9生物传感器介导的检测技术
CRISPR-dCas9系统是将Cas9蛋白的RuvC和NHN两个核酸酶活性区域同时进行了突变,成为剪切功能去除后的突变体,该突变体不具有核酸酶活性但是仍然保留了高特异的识别能力。研究人员已经将这种功能蛋白有效的利用在识别和成像等领域。
韩国蔚山大学医学院Yong Shin教授将CRISPR-dCas9系统、RPA等温扩增技术以及硅微环谐振器(silicon mirroring resonator,SMR)相结合,在硅微环谐振器上完成核酸的检测[15]。为了能够同时扩增和检测核酸,该方法将靶序列特异性引物固定在SMR生物传感器表面,引物在恒定温度条件下进行RPA等温扩增反应(图6)。对于DNA,引物结合重组酶以延伸DNA;对于RNA,通过RT-RPA进行扩增。扩增产物中含有能够被dCas9-sgRNA复合体识别的序列,dCas9与传感器表面上的扩增产物结合并增加传感器表面的分子量,从而通过增加折射率变化来增加检测灵敏度。该方法在20 min内可以完成对靶标的检测,通过对蜱传相关疾病检测应用,可以达到0.54 amol,这种检测灵敏度比RT-PCR检测灵敏度高100倍。
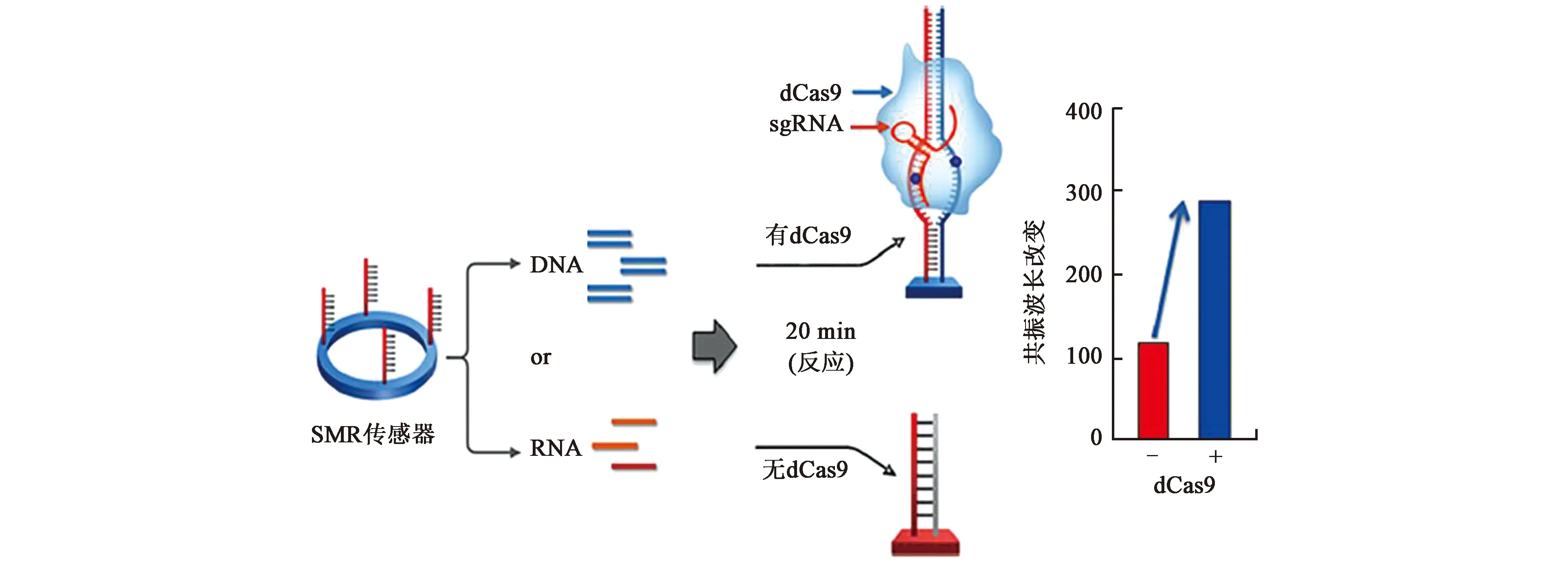
图6 CRISPR介导的SMR传感器原理图[15]Fig.6 The principle of of SMR biosensor mediated by CRISPR[15]
国防科技大学朱凌云团队将CRISPR-dCas9系统与滚环体外核酸扩增技术应用到生物传感器中建立了RCA-CRISPR-split-HRP传感器,实现了对miRNA的快速高效检测[16]。其原理是靶标miRNA促发RCA扩增形成多种发卡状结构,再通过sgRNA-dCas9系统将批量的HRP酶活蛋白重新靠近产生活性,最后通过加入TMB发生显色反应实现对靶标miRNA的检测(图7A)。该方法能够实现fmol级别、单碱基差异的miRNA检测。该团队还利用上述方法制作试剂盒,该试剂盒在实地检验过程中可以在37℃条件下对于血液样品4 h内完成,并且试剂盒保质期可以达到一年(图7B)。
dCas9-sgRNA复合体能够通过碱基互补配对与靶标进行结合,这种结合方式与抗原抗体免疫结合相类似。利用这种识别机制,韩国生物科学与生物技术研究所Juyeon Jung研究团队设计了基于dCas9-sgRNA的核酸荧光原位杂交显色传感器用于耐甲氧西林金黄色葡萄球菌(methicillin-resistantStaphylococcusaureus,MRSA)的检测[17]。体外表达的dCas9一端连接磁性纳米颗粒,通过与sgRNA结合后对MRSA基因组特异性片段识别(反应时间30 min)。结合后,通过磁力进行吸附洗脱,若样品中含有靶标序列,dCas9-sgRNA-靶标-磁珠复合物将吸附停留在检测池,加入DNA双链荧光染料SYBR Green I,通过判断是否产生绿色进行MRSA的可视化检测(图8)。通过测得溶液荧光强度可以进行定量检测,检测限为10 CFU/mL。该方法舍去了常用检测方法中对于靶标序列的扩增过程,有效减少了时间。结果通过颜色变化能够更加直观的体现结果,从而有效的实现MRSA病原菌的现场快速检测。
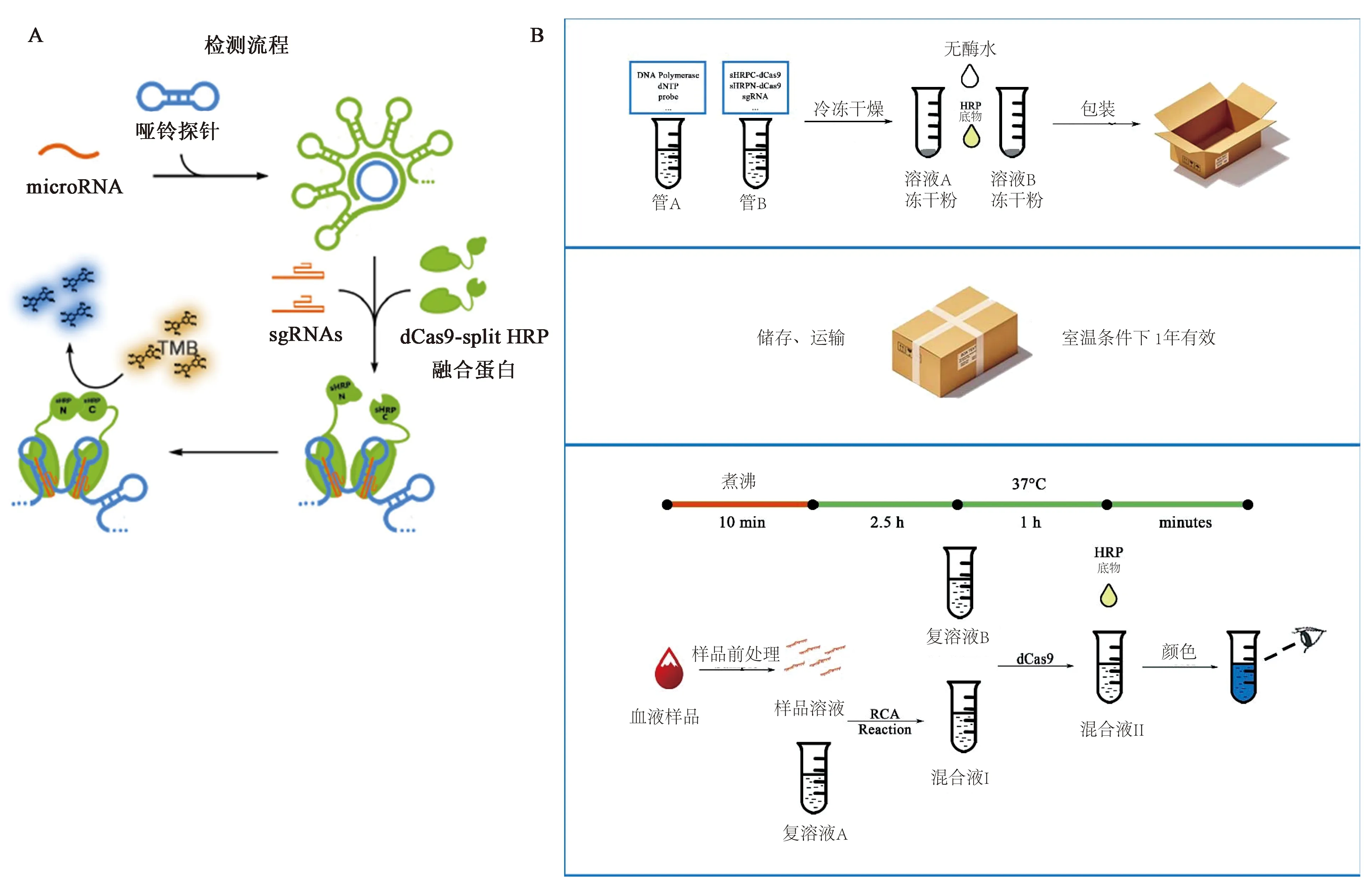
A:CRISPR-Cas9介导的RCA等温扩增生物传感器原理;B:CRISPR-RCA试剂盒制作流程。图7 CRISPR-Cas9介导的RCA等温扩增生物传感器原理与CRISPR-RCA试剂盒制作流程[16]Fig.7 The principle of RCA amplification biosensor mediated by CRISPR and the production progress of CRSIPR-RCA reaction kit[16]
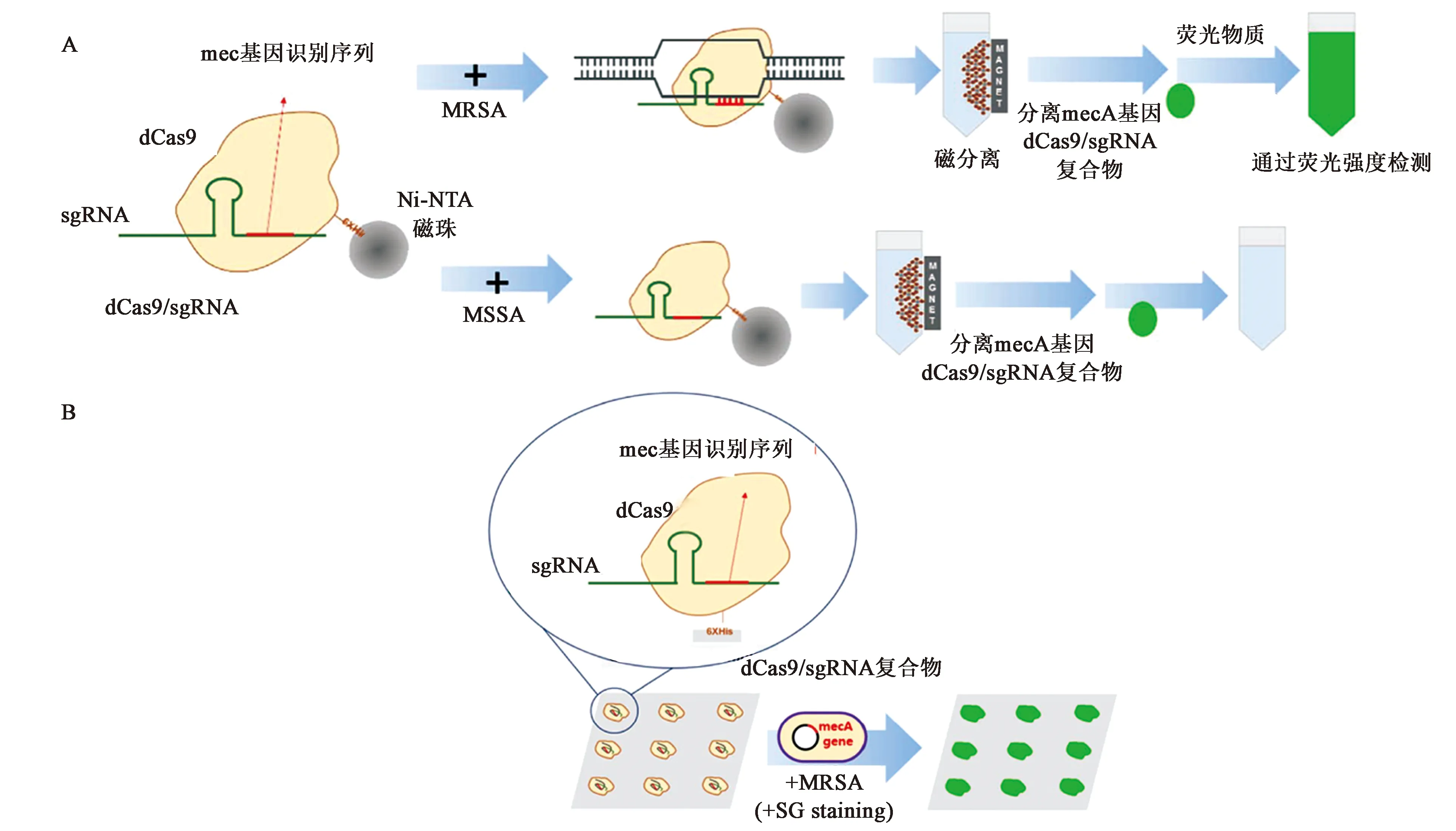
A:CRISPR-dCas9介导FISH方法检测MRSA原理图;B:芯片原位显色方法。图8 CRISPR-dCas9介导FISH方法检测MRSA原理图与芯片原位显色方法Fig.8 The principle of MRSA detection by CRSIPR-dCas9 mediated FISH biosensor and in situ detection method on chip
CRISPR-dCas9系统作为一种核酸特异性识别工具,通常需要对目标进行一定的扩增富集后才能更好的行使作用[18-19]。为了建立更加简便的一步完成靶标识别的方法,研究人员开始引入纳米材料构建方法。纳米材料由于其优越的物理性质,可以通过灵敏的光学性质进行信号输出。荷兰代尔夫特理工大学Kavli纳米科学研究所的Cees Dekker团队建立了基于CRISPR-dCas9的固态纳米孔对DNA进行检测[20]。该方法首先将靶RNA与dCas9预孵育,然后与靶标DNA样品混合(图9A)。使结合CRISPR-dCas9的DNA通过固态纳米孔,其中dCas9蛋白结合的DNA信号能够产生明显的特征,从而鉴定特定的DNA序列(图9B,C)。并且,该团队还验证了在高盐浓度下,该体系仍然适用。由于dCas9蛋白增大了流体动力学半径,使得靶标信号增强,即使在大孔径条件下仍然可以实现靶标检测。该团队成功应用该方法对DNA进行了分型,并且预测该方法可以应用到疾病菌株的快速鉴定、抗生素抗性检测以及基因组的分型中。
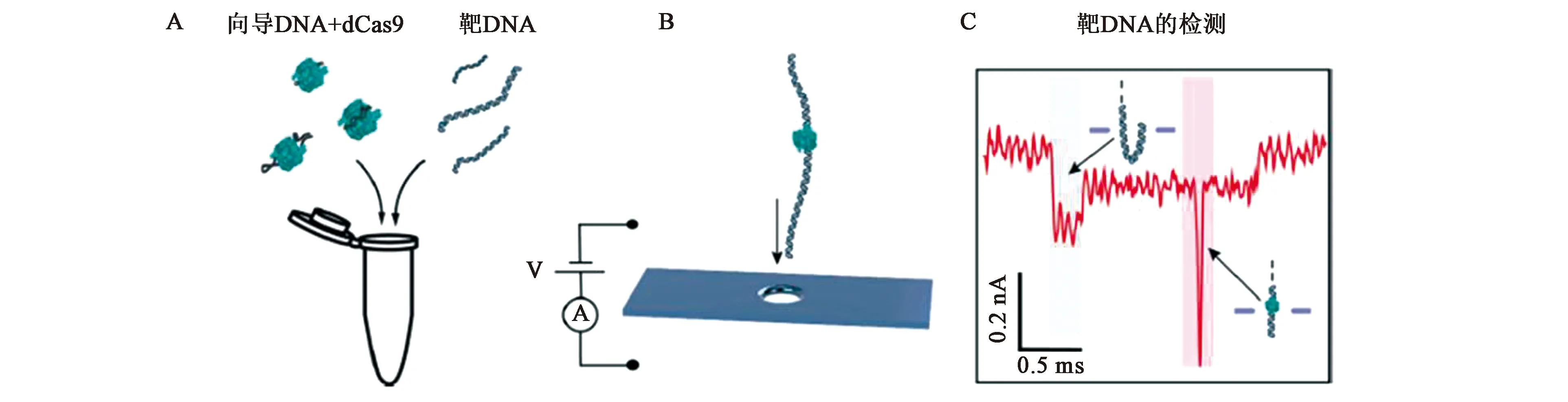
A:CRISPR-sgRNA结合靶DNA;B:CRSIPR识别复合体通过固态纳米孔;C:电流变化实现DNA检测图9 基于CRISPR-dCas9的固态纳米孔检测DNA[20]Fig.9 DNA detection of solid state nanopore biosensor based on CRSIPR-dCas9[20]
4 CRISPR-Cas12a生物传感器介导的检测技术
CRISPR-Cas12a蛋白也叫做Cpf1蛋白,属于第二大类,第V型系统。该蛋白在RNA指导下,能够结合并切割外源DNA,也是细菌免疫系统的组分。RNA引导的Cas12a结合DNA后具有切割任意单链DNA的活性,完全降解ssDNA分子。加州大学伯克利分校Jennifer A.Doudna教授发现Cas12a拥有这种特殊性质之后,通过将Cas12a与等温扩增相结合,创建了一种名为DETECTR(DNA endonuclease targeted CRISPR trans reporter)的方法[21]。首先将血液样本进行前处理,之后进行RPA的初步扩增。再将crRNA-Cas12a复合物以及进行荧光淬灭的ssDNA探针一起加入,当与双链DNA特异性结合之后,活化Cas12a会无差别的切割ssDNA探针,从而释放荧光信号(图10)。该方法实现了对DNA在μmol级的检测,并且DETECTR能够快速、特异地检测到患者样本中的人乳头瘤病毒,为分子诊断提供了一个简单的平台。
5 CRISPR-Cas13a系统介导的检测技术
Cas13a蛋白又叫做C2C2蛋白,属于第二大类的VI型CRISPR-Cas系统,是2015年张锋和Koonin利用RNA测序技术分析得出新一类VI型CRISPR-Cas系统的代表[19]。Cas13a蛋白在与sgRNA形成功能性复合物后,再与靶RNA识别可以在形态学上激活Cas13a的非特异性反式核糖核酸酶活性,能够对任意单链RNA进行剪切。CRISPR-Cas13a系统的出现掀起了针对RNA靶标的新型基因编辑应用浪潮。
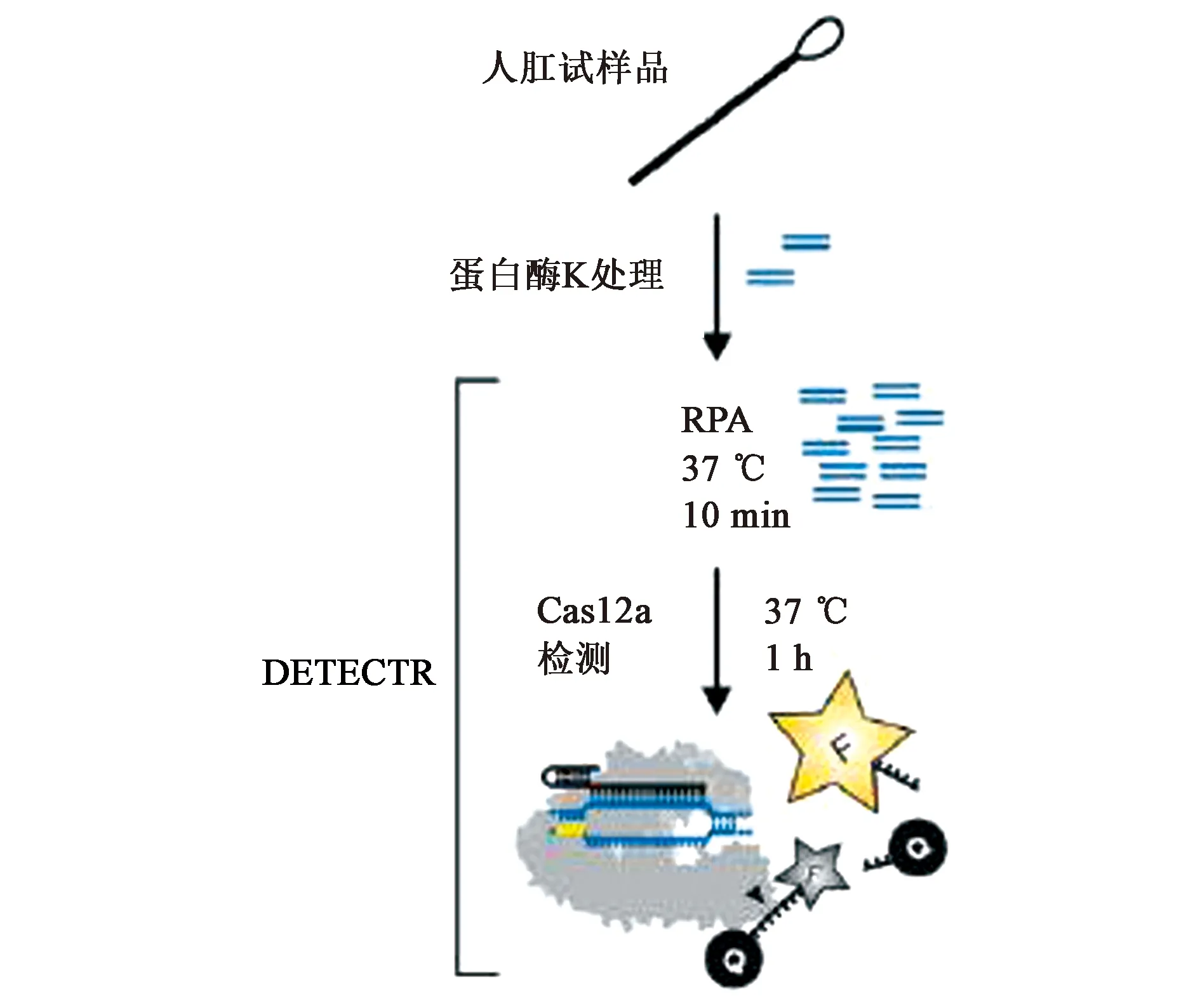
图10 DETECTR检测双链DNA原理图[21]Fig.10 DNA detection principle by DETECTR biosensor[21]
根据以上性质,2017年张锋团队构建了基于CRISPR-Cas13a系统的传感器,称为SHERLOCK(specific high sensitivity enzymatic reporter unlocking),实现了对DNA快速、廉价、高灵敏的检测[19]。由于Cas13a/sgRNA系统识别底物为RNA序列,所以在进行DNA检测时,需要后续进一步转录过程。SHERLOCK技术首先利用了RPA等温扩增技术将样品中的靶标DNA进行富集,经过T7转录法将DNA转录成单链RNA(图11)。将构建好的crRNA-Cas13a复合体以及荧光RNA探针与样品RNA混合,当样品中存在与crRNA碱基互补的序列时,Cas13a的RNase酶活性激活会对周围荧光淬灭RNA探针进行剪切从而释放荧光信号。该团队利用CRISPR-Cas13a传感器实现了体外μmol级别的检测并且可以识别单碱基的错配,同时应用于寨卡病毒和登革热病毒中特定菌株的检测、区分致病菌、人类DNA基因分型,及鉴定无细胞肿瘤DNA突变等。此外,SHERLOCK反应试剂可以冷冻干燥,以实现长期储存,并可在纸上轻松复原,为现场检测应用提供了方法基础。东南大学何农越教授团队利用该技术成功实现了对禽流感H7N9病毒的现场检测[22],在5 min内可以检到1 nmol级的RNA模板,与RT-RPA结合50 min内可以检测到1 fmol级的DNA模板。
鉴于Cas13a对RNA的专属识别机制,利用其建立针对miRNA的检测方法更加准确、方便。邢达教授团队首次利用纤毛菌体内的Cas13a建立了miRNA的检测传感器(图12)[23]。该方法摆脱了对靶标序列预扩增的过程,有效的避免了基因组DNA污染的影响。该方法的高特异性不仅来源于crRNA与模板的碱基互补配对,Cas13a蛋白酶的高保真度使该方法能够区分存在于miRNA末端的单核苷酸突变。利用此方法检测miRNA,检测线可达到4.5 amol级别,并且线性范围跨越4个数量级,从10 amol级到100 fmol级。此外,在面对复杂生物样品(血清)时,该方法也表现出了良好的应用效果。
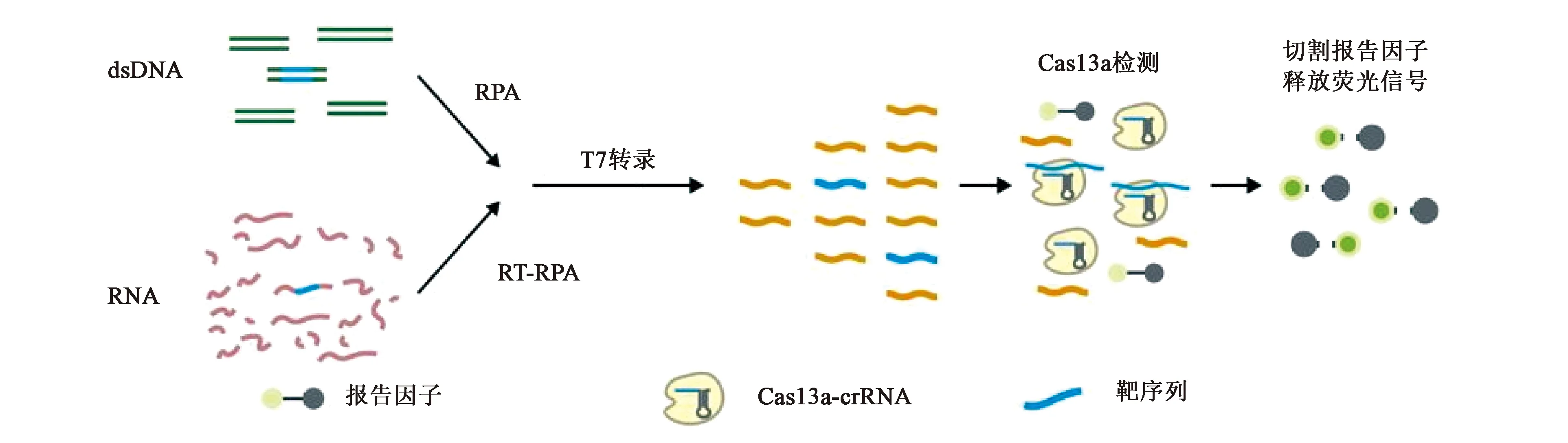
图11 SHERLOCK检测核酸原理图[19]Fig.11 Detection principle of SHERLOCK biosensor[19]
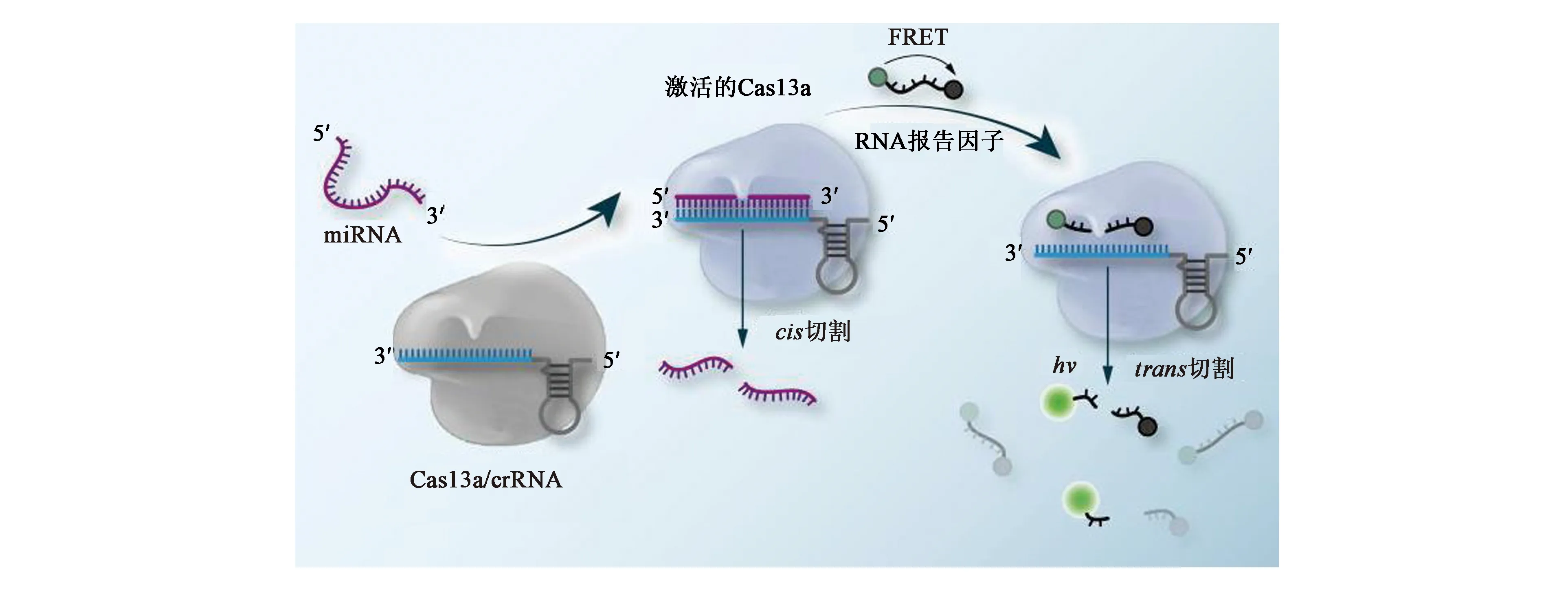
图12 CRISPR-Cas13a检测miRNA原理图[23]Fig.12 miRNA detection principle of CRSIPR-Cas13a biosensor[23]
6 CRISPR-Cas系统在胞内成像中的应用
细胞中,基因组DNA呈现高度折叠状态并存在于三维空间中。细胞核中染色质的空间组织和不同染色质区域的相对位置关系与正常发育和疾病中的基因调控紧密相关。通过对胞内的染色质结构域或基因进行可视化定位,能够进一步用于疾病相关研究和诊断。传统的胞内核酸成像多利用探针杂交技术,例如DNA荧光原位杂交技术。但传统方法需要首先对基因组进行物理或者化学处理,例如通过热和甲酰胺进行严格处理以使dsDNA变性以允许探针杂交,因此该方法存在影响生物结构和基因组组织完整性的风险,并且寡核苷酸探针的高成本也是传统方法的一大缺陷。因此,开发更简单有效、成本低廉和稳健的成像技术具有重要意义。
CRIPSR系统对于核酸序列的特异性结合方式为建立新型识别机制提供了新方法,并且Cas蛋白的生物相容性也为该系统实现胞内成像提供了生物安全保证。核酸酶缺陷型Cas9衍生物(dCas9)常常被用于胞内核酸物质的识别,通过结合不同荧光基团实现胞内物质的实时成像和原位成像。早在2013年,加州大学旧金山分校的黄波教授团队通过优化的CRISPR-Cas系统对活细胞中的基因组基因座进行动态成像[24]。利用EGFP标记的dCas9和结构优化的sgRNA,成功的对端粒中重复元件和活细胞中编码基因进行稳定成像(图13A)。美国霍华德休斯医学研究所的Robert H.Singer研究员团队将此方法进行了改进[25],在dCas9上连接不同荧光基团,实现对胞内基因组两个基因座的原位成像(图13B)。
以上几种方式都是将荧光基团修饰在dCas9蛋白末端,信号的富集主要由胞内基因组上靶标序列的重复性决定。所以上述方法在建立时,都是针对于染色体着丝粒前端多重复系列进行设计成像的,在面对靶标序列为低重复甚至单拷贝时具有很大的局限性。研究人员后续利用CRISPR-dCas9进行了进一步的改良[26-27],通过设计具有特殊发卡结构的sgRNA,将不同荧光蛋白连接到相应发卡位置,能够实现荧光信号的富集,再通过与靶标序列结合,完成胞内核酸序列的成像(图13C,D)。
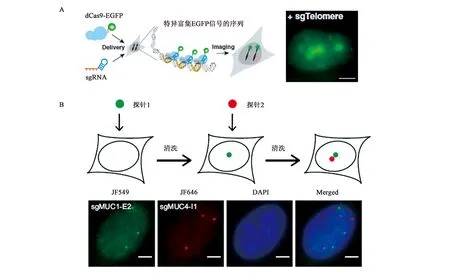
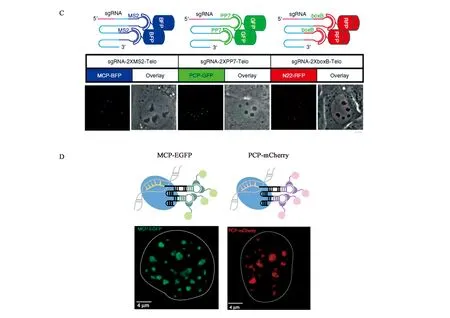
A:EGFP标记的dCas9对端粒中重复序列的准确成像;B:双荧光标记的dCas9实现基因组双基因座成像;C,D:荧光标记sgRNA实现胞内核酸成像图13 基于CRISPR-dCas9技术的胞内核酸成像[24-27]Fig.13 Intracellular nucleic acid imaging by biosensor based on CRSIRP-dCas9[24-27]
7 展望
CRISPR-Cas技术作为当下最常用的基因编辑工具,被广泛的用于新品种培育、基因通路研究以及基因药物研制等领域,并取得了巨大的成功。但是,关于CRISPR技术的负面报道也经常出现,最受关注的即CRSIPR技术的脱靶效应问题。一些报道指出,CIRSPR系统介导的体内编辑基因组DNA双链断裂修复会产生大量脱靶效应,极有可能造成切割位点远端DNA大片段丢失、乃至其他更为复杂的基因突变[28-29]。虽然,有些研究报道后被撤回,但关于CRISPR技术的潜在风险引起了人们对于该技术在医学领域应用的担忧。此外,该技术应用于体内还面临着很多挑战,如编辑效率以及递送方式等问题。但是,毫无疑问的是CRIPSR的时代已经来临,越来越多的研究人员已经投入到此项技术的改进和完善工作中。多种基于新型CRISPR系统的编辑工具逐渐被开发出来。为了避开其潜在的风险,我们可以利用这种新型的技术应用于疾病相关检测、成像领域。通过多种跨学科技术的联用建立生物传感器,将CRIPSR系统的优势发挥到最大。建立多重逻辑关系,通过核酸实现多种疾病标志物的检测,从而在分子水平上实现更高精确度和可靠性的疾病诊断。
猜你喜欢
世界科学技术-中医药现代化(2022年9期)2023-01-17
中国慈善家(2022年3期)2022-06-14
现代苏州(2022年9期)2022-05-26
快乐语文(2021年34期)2022-01-18
今日农业(2021年11期)2021-08-13
军民两用技术与产品(2021年10期)2021-03-16
中国生殖健康(2020年4期)2020-12-09
中国(俄文)(2020年8期)2020-11-23
中西医结合肝病杂志(2020年2期)2020-10-27
世界农药(2019年3期)2019-09-10
