
亚铁烷基咪唑自旋交叉配合物的合成及其金属有机凝胶性能
2019-12-11 05:51王娅琴张海霞张舒恒葛芳圆陈雨欣顾志国
无机化学学报 2019年12期
王娅琴 张海霞 张舒恒 何 威 葛芳圆 陈雨欣 顾志国*,,2
(1江南大学化学与材料工程学院,无锡 214122)
(2合成与生物胶体教育部重点实验室,无锡 214122)
0 引 言
超分子凝胶是一种介于固体和液体之间的准固态物质,它通过小分子间π-π堆积、疏水作用以及氢键等非共价作用力自组装形成三维网络结构,进而阻止溶剂分子的自由流动形成凝胶体系[1]。与以聚合物为凝胶因子的传统凝胶相比,小分子结构简单,且分子间通过弱作用力形成空间网状结构,使其具有更好的生物相容性、刺激响应性和自愈能力[2]。金属有机凝胶(metal-organic gels,MOGs)是指在具有配位能力的有机配体中,引入金属离子,通过金属配位作用诱导形成凝胶;或者以金属配合物为凝胶因子,通过非共价相互作用形成凝胶[3]。金属有机凝胶兼具金属离子和有机配体的特点,其种类的多样性实现了合成上的无限性。金属有机凝胶的配位环境可控,金属中心多样的物理化学性质(如光、磁、氧化还原性等)使其在可视化识别[4]、药物化学[5]、催化[6]等领域具有潜在应用价值。
自旋交叉(spin-crossover,SCO)是指在一定的外界环境干扰(如光照、压力、磁场等)下,电子排布为3dn(4<n<7)的过渡金属配合物的金属中心离子在八面体场中发生高自旋和低自旋相互转换的现象,也称磁双稳态现象[7]。外界环境的干扰导致配合物晶体场发生改变,进而引起金属中心离子的电子态以及配合物的分子结构的变化,这一过程往往伴随着配合物的光学性质、磁性等物理化学性质的改变,使其在显示设备、信息储存等领域有着广阔的应用前景[8]。近年来,具有SCO性能的金属有机凝胶由于其在分子开关、光学器件以及信息存储等领域的潜在应用而受到广泛关注[9]。将具有SCO性能的固体材料应用于凝胶软材料的制备中,在保持SCO性能的基础上,引入凝胶性能得到新型双刺激响应性金属有机凝胶材料,从而有效地拓宽金属配合物的应用范围[10]。
我们利用咪唑亚胺作为配位单元形成的合适配体场,赋予Fe(Ⅱ)超分子体系自旋交叉性能。同时,咪唑环上的含氢氮原子易与卤代烃发生烷基化反应,因此易于在咪唑环上引入长烷基链,使得亚铁烷基咪唑配合物具有凝胶功能。我们通过1-溴烷烃对咪唑-2-甲醛进行烷基化反应,制备了系列1-烷基-1H-咪唑-2-甲醛,并进一步与1-苯乙胺和二价铁金属盐Fe(BF4)2·6H2O反应后得到配合物1~5。受烷基链长度影响,配合物1~5具有不同的磁性质。以具有较长烷基链(n≥14)的配合物2~5作为凝胶因子,与环己烷、正戊醇、DMF、乙腈等不同的有机溶剂混合进行溶剂筛选,最终选定环己烷作为溶剂得到金属有机凝胶MOG2~MOG5,并研究了凝胶的刺激响应性能。
1 实验部分
1.1 仪器与试剂
所用仪器包括FTLA2000-104红外光谱仪,扫描范围4 000~500 cm-1;AVANCEⅢ400 MHz全数字化核磁共振谱仪;Elementar Corporation Vario ELⅢ元素分析仪;SMART APEXⅡDUO CCD单晶衍射仪;MPMS-XL7 SQUID磁测量仪;DHR-2流变仪;S-4800场发射扫描电子显微镜(操作电压2 kV)。所用试剂中1-苯乙胺、六水合四氟硼酸亚铁、乙腈、乙醚均为分析纯。1-庚烷基-1H-咪唑-2-甲醛、1-十四烷基-1H-咪唑-2-甲醛、1-十六烷基-1H-咪唑-2-甲醛、1-十八烷基-1H-咪唑-2-甲醛、1-二十烷基-1H-咪唑-2-甲醛的合成见Supporting information。
1.2 配合物1~5的合成
1.2.1 配合物1的合成
在50 mL的三颈烧瓶中,加入1-庚烷基-1H-咪唑-2-甲醛(1 mmol,0.194 g)、1-苯乙胺(1 mmol,0.121 g)和20 mL乙腈。氮气保护下,混合物在80℃搅拌回流2 h,得浅黄色溶液。将反应液冷却至室温并加入Fe(BF4)2·6H2O(0.33 mmol,0.112 g),溶液立即变成紫红色。此混合溶液在室温下继续搅拌1 h,过滤除去杂质,得深紫色滤液。将滤液转移至试管放于广口瓶中,以无水乙醚为不良溶剂,扩散法培养晶体。3 d后,得到紫红色块状晶体,产率:41.5%。C57H81B2F8FeN9元素分析计算值(%):C 61.03;H 7.28;N 11.24;实验值(%):C 60.98;H 7.38;N 11.14。IR(KBr,cm-1):2 923,2 852,1 616,1 488,1 465,1 450,1 380,1 307,1 091,763,702,622(Supporting information,图S7)。
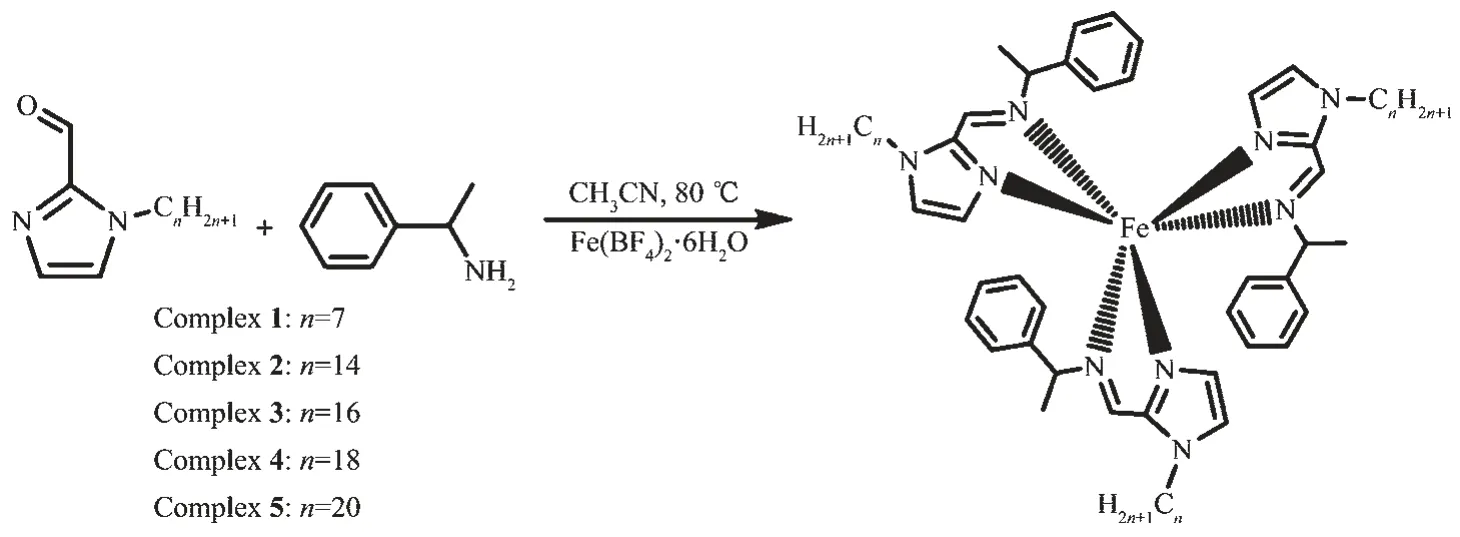
图1 配合物1~5的合成示意图Fig.1 Synthetic routes of complexes 1~5
1.2.2 配合物2~5的合成
参考配合物1的合成步骤,分别用1-十四烷基-1H-咪唑-2-甲醛、1-十六烷基-1H-咪唑-2-甲醛、1-十八烷基-1H-咪唑-2-甲醛、1-二十烷基-1H-咪唑-2-甲醛和1-苯乙胺与Fe(BF4)2·6H2O合成配合物2~5,最终产物均为橙色粉末。
配合物2产率:46%。C78H123B2F8FeN9元素分析计 算 值(%):C 66.15;H 8.75;N 8.90;实 验 值(%):C 65.66;H 8.76;N 8.91。IR(KBr,cm-1):2 918,2 850,1 645,1 618,1 488,1 467,1 454,1 377,1 305,1 083,763,721,702(图S7)。
配合物3产率:52%。C84H135B2F8FeN9元素分析计 算 值(%):C 67.24;H 9.07;N 8.40;实 验 值(%):C 67.13;H 9.19;N 8.33。IR(KBr,cm-1):2 920,2 850,1 647,1 616,1 488,1 467,1 450,1 377,1 299,1 083,761,721,702(图S7)。
配合物4产率:43%。C90H147B2F8FeN9元素分析计 算 值(%):C 68.21;H 9.35;N 7.96;实 验 值(%):C 68.15;H 9.47;N 8.23。IR(KBr,cm-1):2 918,2 850,1616,1490,1469,1379,1305,1091,966,916,763,703,624(图S7)。
配合物5产率:48%。C96H159B2F8FeN9元素分析计 算 值(%):C 69.09;H 9.60;N 7.55;实 验 值(%):C 68.95;H 9.72;N 7.78。IR(KBr,cm-1):2 916,2 850,1 638,1 616,1 490,1 469,1 379,1 299,1 083,763,721,703(图S7)。
1.3 X射线单晶衍射
将配合物1在173(2)K温度下置于SMART APEXⅡDUO CCD衍射仪上,用石墨单色化的Mo Kα射线(λ=0.071 073 nm),以ω扫描方式收集数据,用SMART软件还原晶胞参数,并用SAINT软件[11]对数据进行精修。实验过程中设置扫描宽度ω为0.30°,剩余大量数据集采用SAINT消减或修正洛仑兹和极化效应消减,用SADABS软件进行衍射强度矫正[12]。晶体结构用SHELXS-97程序采用直接法解出并基于F2用SHELXS-97程序对所有非氢原子坐标及其各项异性参数进行了全矩阵最小二乘法精修[13]。主要晶体学数据列于表1,主要键长与键角列于表S1。
CCDC:1920725,1。
2 结果与讨论
2.1 配合物的表征与性能测试
2.1.1 配合物1~5的合成与表征
通过不同链长的1-烷基-1H-咪唑-2-甲醛、1-苯乙胺和四氟硼酸亚铁采用“一锅法”组装合成配合物1~5。红外图谱分析表明,2 923~2 850 cm-1范围出现的特征吸收峰为甲基和亚甲基上饱和C-H键的伸缩振动峰,在1 647~1 616 cm-1范围内出现的C=N双键伸缩振动吸收峰且1 690 cm-1左右中等强度的C=O双键伸缩振动吸收峰消失,1 460 cm-1左右出现了芳环骨架振动吸收峰,在1 090~1 080 cm-1范围出现阴离子特征吸收峰(图S2、S7),表明亚铁烷基咪唑配合物的形成。配合物1~5元素分析实验值与计算值基本吻合,进一步证明目标配合物的合成。
2.1.2 配合物1晶体结构分析

表1 配合物1的晶体数据Table 1 Crystallographic data for complex 1
为了直观说明亚铁烷基咪唑配合物的结构,我们测试了配合物1的X射线单晶结构,晶体结构信息和键长键角见表1及Supporting information。如图2所示,X射线衍射结果显示配合物1中,每一个结构单元由[Fe(L1)3]2+阳离子和2个BF4-抗衡阴离子组成单核金属配合物。配合物1中每个Fe(Ⅱ)金属中心与来自3个配体的6个氮原子配位形成变形八面体。Fe(Ⅱ)-N键的平均键长为0.197 5 nm,说明温度为173 K时,Fe(Ⅱ)中心处于低自旋状态[14]。在配合物1中,C44-C49、C13-C18、C32-C37组成3个苯环π平面,质心分别为Cg1、Cg3、Cg5;C1-C3与N1-N2、C20-C22与N4-N5、C39-C41与N7-N8组成3个咪唑环π平面,质心分别为Cg2、Cg4、Cg6。每一个苯环与相邻配体上的咪唑环之间存在分子内π-π作用,其面心距离分布在0.359 6~0.405 9 nm范围内。分子内π-π相互作用对配合物超分子结构起到了稳定作用。
如图3a所示,每个[Fe(L1)3]2+阳离子单元中烷基链末位上的C-H与相邻配合物咪唑环形成的分子间C-H…π相互作用;同时苯环邻位上的C-H与相邻配合物苯环形成分子间C-H…π相互作用。这2种C-H…π作用距离长度分别为0.283 5和0.359 5 nm。每个[Fe(L1)3]2+阳离子单元与周围相邻的4个阳离子单元通过C-H…π作用形成二维超分子网络结构。如果将每个[Fe(L1)3]2+阳离子看作1个节点,分子间C-H…π作用看作1个连接体,那么这个超分子结构可以看作是一个44网络拓扑结构(图3b)。配合物1中分子间C-H…π作用进一步稳固了晶体的堆积结构。
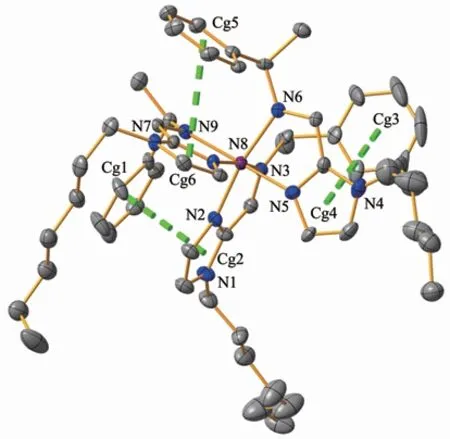
图2 配合物1的椭球率30%的晶体结构图Fig.2 ORTEPdiagram showing the structure of complex 1 with the atom numbering scheme at 30%probability levels
2.1.3 磁性研究
在100℃温度下,对样品进行12 h的干燥处理,以去除配合物中所有易挥发结晶溶剂分子。在1 000 Oe的外加磁场下,利用MPMS-XL7 SQUID磁测量仪以3 K·min-1的速率收集配合物1~5在100~400 K温度范围内降温的磁性数据,摩尔磁化率(χM)计算公式:χM=Mm/(Hms),其中M为分子量;m为磁矩,单位为emu;H为磁场强度,单位为Oe;ms为样品质量,单位为g。
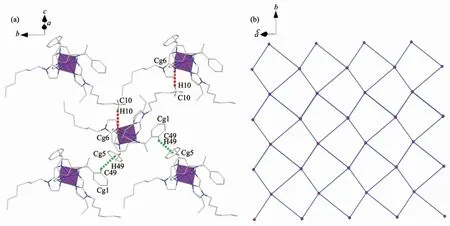
图3 (a)配合物1的二维C-H…π超分子结构;(b)配合物1的44超分子网络拓扑结构Fig.3 (a)2D C-H…πsupramolecular network structure of complex 1;(b)View of the 44 supramolecular network topology for complex 1
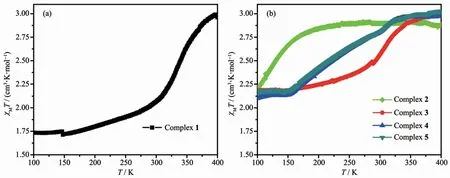
图4 (a)配合物1的变温磁化率曲线图;(b)配合物2~5的变温磁化率曲线图Fig.4 (a)Variable-temperature magnetic susceptibility measurements for complex 1;(b)Variable-temperature magnetic susceptibility measurements for complexes 2~5
如图4a所示,配合物1表现出不完全的自旋转换行为,自旋转换温度为341 K。配合物1在400 K时磁化率为2.99 cm3·K·mol-1,温度降低至300 K时,磁化率快速降为2.07 cm3·K·mol-1。继续降温时,磁化率缓慢降低至100 K时的1.73 cm3·K·mol-1。如图4b所示,由于烷基链长度的不同,配合物2~5表现出不同的磁性质。配合物2在400 K时磁化率为2.87 cm3·K·mol-1,温度降至180 K,磁化率基本保持不变,表明配合物2为高自旋顺磁性化合物[15]。温度继续降低,配合物2的磁化率迅速降低至100 K的2.24 cm3·K·mol-1。配合物3在400 K磁化率为2.99 cm3·K·mol-1,325~150 K温 度 区 间 磁 化 率 由2.79 cm3·K·mol-1降为2.18 cm3·K·mol-1,随后温度降至100 K,磁化率基本保持不变。配合物4和5有着相似的变温磁化率曲线,400~325 K温度区间内磁化率基本保持不变。温度继续降低,配合物4和5发生缓慢的不完全自旋交叉行为,分别从325 K的2.93和2.92 cm3·K·mol-1降为150 K的2.14和2.18 cm3·K·mol-1,转变温度分别为230和233 K。磁性数据表明,烷基链长度对配合物1~5的磁性有着显著的影响。众所周知,晶体堆积方式和分子间作用力对Fe(Ⅱ)配合物的自旋转换行为有着复杂的影响,而随碳链的增长配合物中烷基链间的范德华力增强[16]。由此推测,配合物1~5的分子间作用力存在差异,从而导致配合物1~5表现出不同的磁性质。我们试图测试2~5的晶体结构,从分子角度探讨烷基链长度对磁性的影响,然而由于碳链较长导致配合物衍射较弱,虽然尝试多种培养单晶的方法,最终仍未能得到配合物2~5的晶体结构。
2.2 金属有机凝胶的表征与性能测试
2.2.1 溶剂筛选
分别称取0.05 g配合物2~5置于4个5 mL玻璃瓶中,均加入1 mL有机溶剂,利用振荡、超声或加热的方式使固体充分溶解,冷却后静置10 min得到红棕色物质。倒置玻璃瓶观察瓶中溶液是否流动,若瓶中的液体不具有流动性,则判定凝胶形成。并通过此方法筛选出能够形成凝胶的有机溶剂。不同极性有机溶剂的凝胶形成情况见表2。
表2结果显示,配合物2~5在DMF、乙腈、异戊醇和异辛醇溶剂中,难以形成凝胶。使用异丙醇与乙腈、氯仿的混合物作为溶剂同样无法形成凝胶。环己烷、正戊醇、正己醇、正庚醇、环己醇和正辛醇溶剂均能与配合物2~5形成凝胶或部分凝胶。其中,环己烷作为溶剂时,配合物2~5的凝胶效果最好。由此推测凝胶因子在有机溶剂中的凝胶情况与有机溶剂的极性有关。DMF、乙腈、异戊醇和异辛醇溶剂由于极性较高,能够很好地溶解配合物而无法形成凝胶。而非极性的环己烷对配合物2~5的溶解度适中,有利于凝胶的形成。

表2 不同溶剂中配合物2~5的凝胶情况Table 2 Gel conditions of complexes 2~5 in different solvents
2.2.2 SEM表征
以环己烷为溶剂,配合物质量分数为6.4%制备得到金属有机凝胶MOG2~MOG5。由于MOG2~MOG5形貌相似,图5仅展示MOG3样品的SEM图。图5a~b中可以看到MOG3在微观上是由类绸缎片状结构相互交织形成的三维多孔结构,孔隙分布较均匀且连通性好。凝胶因子在形成这样的3D网络互穿结构后,将溶剂分子“锁”在孔隙结构中,形成凝胶。当放大倍数为60 000、90 000时(图3c~d),可以看到类绸缎片状结构表面凹凸不平,且略呈现出条状纹理。
2.2.3 流变学测试
使用美国TA仪器公司DHR-2流变仪,锥板2°角,在(20±0.1)℃下对MOG2~MOG5进行力学测试,测量间距为48μm。首先在施加0.5%应变的情况下,在0.1~100 rad·s-1频率范围内进行频率扫描,再以固定频率对凝胶进行压力应变扫描,结果如图6所示。

图5 MOG3的SEM图Fig.5 SEM images of MOG3

图6 MOG2(a),MOG3(b),MOG4(c)和MOG5(d)的G′,G″随频率变化图Fig.6 G′and G″of metallogels as a function of frequency for MOG2(a),MOG3(b),MOG4(c)and MOG5(d)
由图6可以看到,MOG2~MOG5在0.1~100 rad·s-1的频率范围内,储能模量(G′)始终大于损耗模量(G″),表明弹性性能占主导地位,表现为凝胶特征。随着频率的增大,MOG2~MOG5的G′与G″随频率的增加均有所增大,但G′的增幅大于G″,体系储存能量的能力逐渐提高[17]。此外,以固定频率6.28 rad·s-1对金属有机凝胶进行压力扫描(图S8)。MOG2~MOG5的储能模量(G′)的起始数值大于损耗模量(G″),这是凝胶的一个典型特征[18]。MOG2~MOG5储能模量(G′)开始下降的应变分别为4%、2%、2%、4%,此时凝胶局部解体而流动性增加,应变高于临界应变后,储能模量迅速下降,当MOG2~MOG5的应变分别高于10%、8%、4.5%、19%时,储能模量(G′)低于损耗模量(G″),凝胶体系3D网络结构被破坏,凝胶完全转变为粘性溶液[19]。
2.2.4 刺激响应特性测试
MOG2~MOG5快速振荡或超声处理2 min后,凝胶能迅速变为溶胶;静置10 min后,溶胶失去流动性,重新恢复为凝胶状态,说明该系列金属有机凝胶具有机械响应特性。MOG2~MOG5凝胶在50℃加热5 min后,流动性骤增变为溶胶。在室温条件下冷却,溶胶的流动性迅速下降,15 min后恢复凝胶状态,实现了溶胶-凝胶的相转变,说明该系列金属有机凝胶具有很好的温度响应特性。由此说明,我们成功合成了多刺激响应型金属有机凝胶。由于MOG2~MOG5的刺激响应测试结果相同,图7仅展示MOG4刺激响应测试结果。
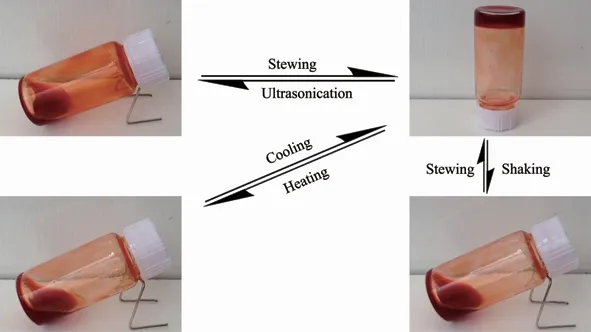
图7 MOG4由于温度、超声处理和剪切应力发生的可逆凝胶-溶胶转变Fig.7 Sol-gel phase transition process of MOG4 stimulated by temperature,ultrasound and shearing strength
2.2.5 凝胶形成机理探讨
由配合物1的晶体结构分析可推测,随着烷基链的增长,相邻配合物之间烷基链的相互交错现象更明显。通过烷基链上H原子与相邻配合物上的咪唑环的C-H…π作用、苯环上H原子与相邻配合物上的苯环的C-H…π作用使得配合物分子间产生紧密联系,为金属有机凝胶的形成提供前提条件。此外,其他非共价作用力(如范德华力、疏水作用)与CH…π作用协同加和,从而促使配合物2~5在有机溶剂中自发组装成3D网络结构,“锁”住溶剂分子进而形成金属有机凝胶。当凝胶因子中取代烷基链足够长时,它所形成的凝胶能对外界刺激(如振荡或者加热)做出响应,一旦外界刺激消失,凝胶能够恢复初始状态,表现出自修复能力[20]。
3 结 论
利用Fe(Ⅱ)和咪唑醛衍生物设计合成得到具有SCO性能的金属有机配合物2~5,并以其为凝胶因子成功得到系列金属有机凝胶。流变学及多项响应测试结果表明,凝胶MOG2~MOG5表现出良好的刺激响应性和自修复能力。基于配合物1晶体结构研究,对该系列金属有机凝胶的形成机理进行了探讨研究。我们成功合成了兼具SCO性能以及凝胶性能的金属有机配合物,为设计和制备新型SCO多功能超分子材料提供了新的思路和途径。
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
云南化工(2022年4期)2022-05-17
作文周刊·八年级版(2020年8期)2020-05-25
黄河黄土黄种人(2019年11期)2019-12-20
健康博览(2019年1期)2019-04-08
科技资讯(2018年16期)2018-10-26
科技视界(2016年19期)2017-05-18
教育(2016年49期)2017-03-20
科技视界(2016年26期)2016-12-17
润滑油(2015年4期)2015-11-20
分析化学(2015年4期)2015-06-08
