
三种酰腙类Schiff碱的锌配合物的合成、晶体结构与表征
2019-12-11 05:50解庆范陈延民
无机化学学报 2019年12期
解庆范 陈延民
(泉州师范学院化工与材料学院,泉州 362000)
锌普遍存在于动植物体内,是多种酶的活性中心,在生理活动中起着重要的作用;许多锌配合物具有抑菌、抗肿瘤等活性。因此,锌配合物的设计与合成一直是生物无机化学备受关注的研究领域[1-4]。酰腙类Schiff碱热稳定性高,它的配位环境与生物体相似,与金属有很强的配位能力,形成的配合物往往呈现比配体更强的生物活性[5-6];其配位方式具有多样性,既能以烯醇式配位,也可以酮式配位,通过对结构的修饰,可以获得结构和性质各异的配合物[7-8]。酰腙Schiff碱的配位方式与金属离子的特征、辅助配体和反应介质等因素有关,尤其是酰腙自身的结构起着关键的作用。酰腙的合成方法简单,可以由酰肼与醛或酮的衍生物直接通过亲核加成反应获得。为了进一步探讨酰腙的配位方式,我们制备了3种酰腙的锌配合物[Zn(Lss)(phen)(DMF)](1)、{[Zn(HLdis)]2·2CH3OH}n(2)和[Zn(Baf)2·CH3OH](3)。酰腙配体H3Ldis和H2Lss的结构如Scheme 1所示。然而,我们试图按Scheme 2所示的路线用Ⅰ和Ⅱ合成酰腙化合物Ⅲ时,得到的却是吡唑啉类的化合物Pzl,也就是说化合物Ⅲ中的次氨基进一步与分子内的苯甲酰基的羰基发生亲核加成反应,从而发生成环化作用。有趣的是,Pzl在与锌离子作用时,五元环重新打开,转变成中间体Ⅲ,进而分子发生重排以单烯醇式(HBaf)与锌离子配位。这种“开-合”转换的现象较为少见。本文主要报导3种配合物和化合物Pzl的晶体结构,其性质有待进一步研究。
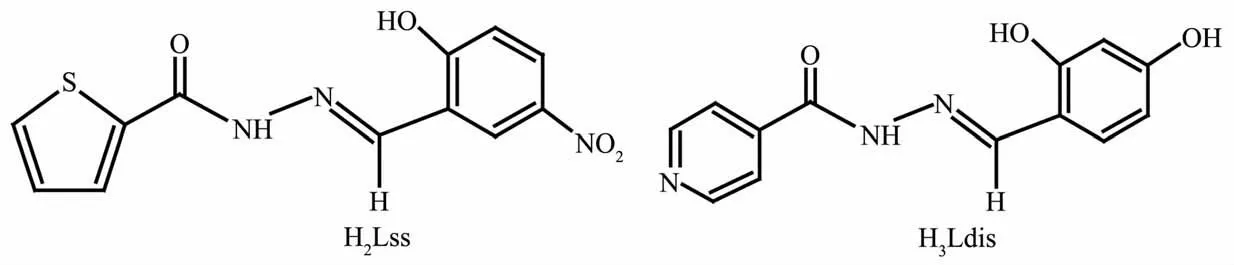
Scheme 1
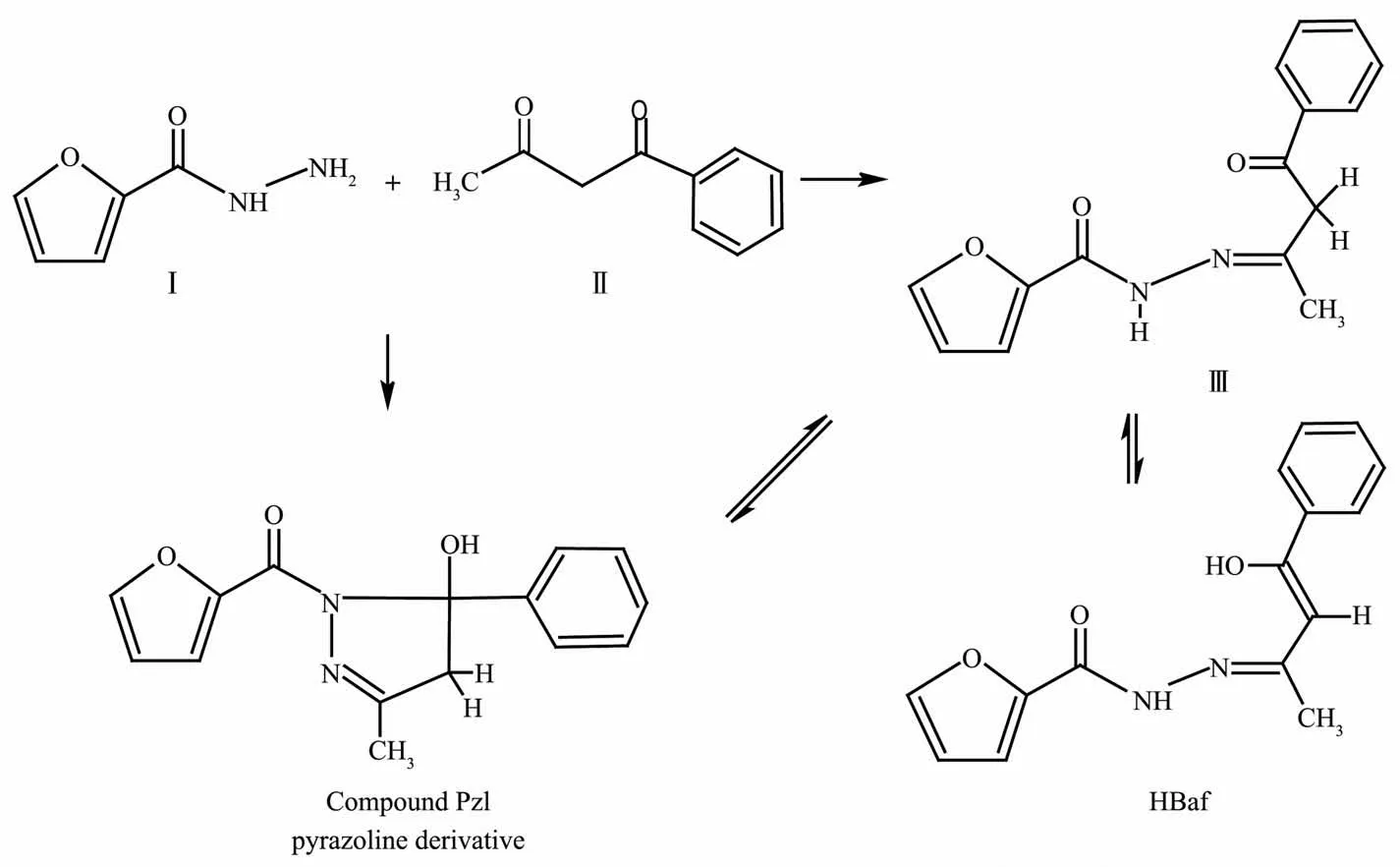
Scheme 2
1 实验部分
1.1 仪器与试剂
所用仪器有德国Elmentar Vario EL元素分析仪,美国Nicolet is10型FT-IR红外光谱仪,上海美普达UV-1800PC型紫外-可见分光光度计,德国塞驰STA 409 PC型综合热分析仪,日本理学Rigaku Saturn724 CCD单晶衍射仪。
所用试剂均为市售分析纯试剂。
1.2 配合物的合成
1.2.1 配合物1的合成
将0.2 mmol乙酸锌、0.2 mmol菲咯啉和0.2 mmol自制的H2Lss[6]溶解于20 mL甲醇和5 mL DMF中,于80℃下加热回流2 h,析出黄色粉末产物;产物用DMF重结晶,滤液于室温下静置3 d后析出淡黄色块状单晶。对C27H22N6O5SZn元素分析的实测值(括号内为理论值,%):C 53.30(53.34);H 3.61(3.65);N 13.71(13.82)。IR(KBr压片,cm-1):2 930w,2 849w,1 655vs,1 614s,1 588m,1 544w,1 505s,1 424s,1 329m,1 291vs,1 218m,1 100m,1 036w,946w,854w,730m,705w。
1.2.2 配合物2的合成
将0.1 mmol自制的H3Ldis[9]溶解于5 mL DMF并置于试管底层,5 mL DMF和甲醇混合溶剂(1∶1,V/V)置于试管的中间层,5 mL含0.1 mmol乙酸锌的甲醇溶液置于上层,静置1周后获得黄色块状晶体。对C14H13N3O4Zn元素分析的实测值(括号内为理论 值,%):C 47.60(47.68);H 3.67(3.72);N 11.81(11.92)。IR(KBr压片,cm-1):3 411(w),1 604(vs),1 547(s),1 516(s),1 445(m),1 342(m),1 319(m),1 295(w),1 217(s),1 128(m),1 027(m),987(m),856(m),758(s),715(s)。
1.2.3 配合物3的合成
(1)化合物Pzl的制备:10 mmol呋喃甲酰肼与10 mmol苯甲酰丙酮在50 mL无水乙醇中回流反应3 h后,自然冷却缓慢挥发,析出无色透明棒状晶体,产率79%。对C15H14N2O3元素分析的实测值(括号内为理论值,%):C 66.51(66.66);H 5.37(5.22);N 10.44(10.36)。IR(KBr压片,cm-1):3 409s,3 099s,2 917w,2 844w,1 634s,1 610vs,1 514s,1 445vs,1 413 s,1 377s,1 313s,1 210m,1 103s,1 077s,1 033m,952w,820s,750vs,703m。
(2)配合物3的合成:1 mmol Pzl和1 mmol乙酸锌溶于10 mL甲醇,于60℃下水浴加热搅拌回流1.5 h,冷却,过滤。滤液密封静置1周后析出亮黄色针状晶体。对C31H30N4O7Zn元素分析的实测值(括号内为理论值,%):C 58.63(58.55);H 4.68(4.76);N 8.89(8.81)。IR(KBr压 片,cm-1):3 403s,3 133vs,1 628s,1 593s,1 526s,1 457s,1 401vs,1 180m,1 122 w,1 072w,1 033w,931w,766w,712w。
1.3 晶体结构的测试
选取尺寸分别为0.50 mm×0.33 mm×0.29 mm、0.22 mm×0.20 mm×0.18 mm和0.45 mm×0.28 mm×0.15 mm的配合物1~3和0.44 mm×0.32 mm×0.29 mm的化合物Pzl的单晶,置于Rigaku Saturn724 CCD单晶衍射仪上,用经石墨单色器单色化的Mo Kα射线(λ=0.071 073 nm)作为X射线源,以ω扫描方式在一定的θ范围内收集单晶衍射数据,其中I>2σ(I)的可观察点用于结构修正。全部衍射强度数据均经Lp因子校正,并进行了经验吸收校正,晶体结构由直接法解出,对全部非氢原子坐标及其各向异性热参数进行全矩阵最小二乘法修正,氢原子坐标由理论计算确定,结构采用SHELXS-97解析,精修采用SHELXL-97程序包[10]完成。晶体学数据详见表1。主要键长和键角列于表2。
CCDC:1465235,1;1404501,2;1516696,3;1516192,Pzl。

表1 化合物的晶体学数据Table 1 Crystallographic data and structure refinement parameters for the compounds
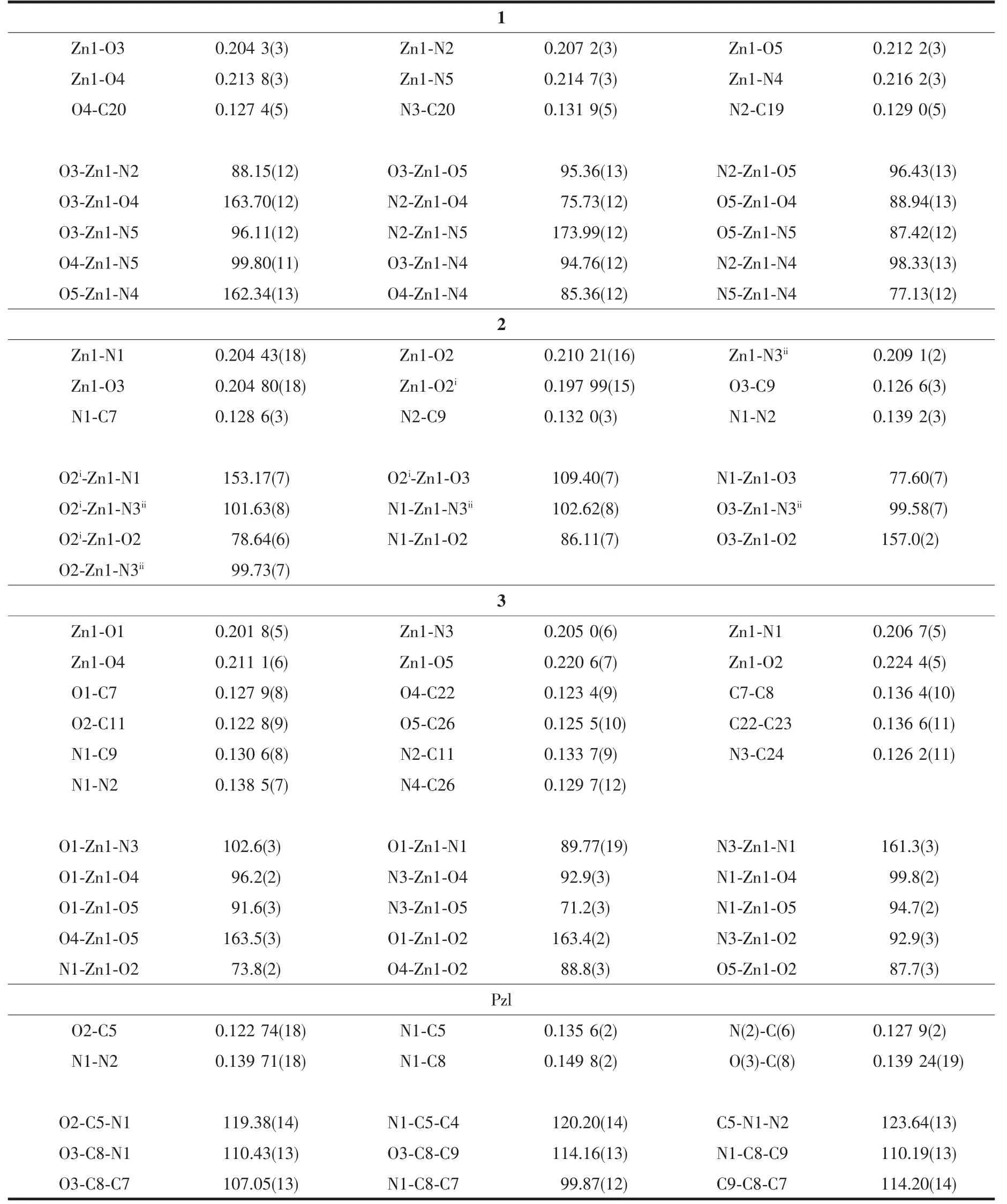
表2 配合物1~3和化合物Pzl的主要键长及键角Table 2 Selected bond lengths(nm)and bond angles(°)for 1~3 and compound Pzl
2 结果与讨论
2.1 晶体结构
2.1.1 配合物1的晶体结构
配合物1属三斜晶系P1空间群,为六配位的单核配合物,分子结构见图1。配合物1由1个中心离子Zn(Ⅱ)、1个酰腙配体阴离子Lss2-、1个菲咯啉phen和1个N,N-二甲基甲酰胺(DMF)组成,Lss2-的酚盐氧原子O3、亚胺基的氮原子N2和羰基氧原子O4、phen的N4和N5以及DMF的羰基O5形成略有畸变的八面体配位环境。键角为75.73(12)°~99.80(11)°和162.34(13)°~173.99(12)°,Zn-N和Zn-O键长为0.204 3(3)~0.214 7(3)nm。N2和N5位于轴向;O3、O4、N4和O5位于赤道,扭转角为21.84°。在2中酰腙配体以烯醇式去质子化形式与Zn(Ⅱ)配位,Lss2-的酰腙片段与水杨基之间呈现较强的共平面性;由于共轭作用N-N呈现出双键的特征,N-N键长为0.138 7(5)nm。

图1 配合物1的30%椭球概率的分子结构图Fig.1 Molecular structure of complex 1 with 30%probability ellipsoids
2.1.2 配合物2的晶体结构
配合物2晶体属单斜晶系P21/n空间群,是一种网箱状的配位聚合物,不对称结构基元见图2。Zn(Ⅱ)处于四方锥配位环境,锥底的4个配原子来自HLdis2-的亚胺基的氮原子N1、酚盐氧原子O2、酰基(烯醇式去质子化)的氧原子O3和相邻配体HLdis2-的O2i,键角为77.65(12)°~156.97(12)°,Zn1-N1键长为0.2040(3)nm,Zn-O的键长为0.1985(3)~0.2106(3)nm;4个配原子几乎在同一平面内,O3-N1-O2-O2i的扭转角为3.78°。锥顶的配原子来自另一相邻配体的吡啶环的氮原子N3ii,Zn1-N3ii键长为0.2087(3)nm,N3ii与锥底形成的键角在99.12(15)°~102.69(14)°范围。
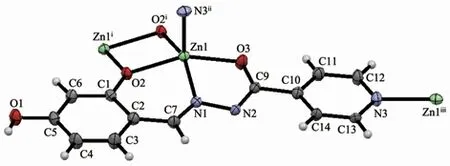
图2 配合物2的椭球率30%不对称结构单元图Fig.2 Asymmetric structure unit of complex 2 with 30%probability ellipsoids
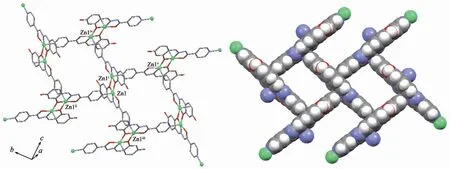
图3 配合物2的二维网箱结构Fig.3 Two-dimensional net cage caption structure of complex 2
水杨醛类Schiff碱有一重要特点,其酚羟基氧原子既能单齿配位,也能单齿桥联配位,这种灵活的配位方式为构建多核配合物或配位聚合物提供了条件。2中酚羟基氧原子O2以μ桥联方式同时与2个Zn(Ⅱ)配位,从而形成一种具有{Zn2O2}簇的中心对称的双核结构基元(图3),Zn(Ⅱ)…Zn(Ⅱ)i间距0.315 9 nm,除Zn(Ⅱ)外双核基元的所有非氢原子几乎完全共平面。由于异烟酰腙吡啶基的存在,使得双核结构基元成为二级建筑块,通过吡啶基N3的桥联作用“砌成”了一种1.074 3 nm×0.843 3 nm的网箱状的二维多孔配位聚合物。这种结构与我们曾报导的镉的配合物[Cd2(μ3-HLdis)2(CH3OH)2]n[9]的结构很相似,不同的是在镉的配合物中甲醇分子参加配位,而锌的配合物中甲醇未与Zn(Ⅱ)配位,而是以游离态形式存在于网箱中。
2.1.3 配合物3的晶体结构
呋喃甲酰肼与苯甲酰丙酮反应并未直接得到预期的酰腙,而是吡唑啉类的衍生物Pzl(图4a)。C6-N2键长0.127 9(2)nm,C5-O2键长0.122 74(18)nm,属典型双键;N1与亚胺基和羰基p-π共轭的结果使得N1-C5、N1-N2比单键键长短得多,键长分别为0.1356(2)和0.139 71(18)nm。N1-C8、C6-C7和C7-C8则为典型的单键,键长分别为0.149 8(2)、0.148 7(3)和1.537(2)nm。呋喃环、羰基和吡唑啉的所有原子共平面,与苯环平面呈88°的夹角。2个分子通过羟基与羰基之间的氢键(O3-H3…O2i)形成氢键二聚体,O3…O2i和H3…O2i(Symmetry codes:i1-x,1-y,1-z)间距分别为0.278 6和0.202 6 nm,∠O3-H3…O2i为153.95°。作为配合物3的前驱体,Pzl在与Zn(Ⅱ)作用时N1-C8键断开发生分子重排,以苯甲酰基丙酮缩呋喃甲酰腙(HBaf)的结构与Zn(Ⅱ)配位。
配合物3由2个非对称性的Baf-和1个Zn(Ⅱ)组成,4个羰基氧原子O1、O2、O4和O5与2个亚胺基氮原子N1和N3构成畸变八面体配位环境,键角在71.2(3)°~102.6(3)°和161.3(3)°~163.5(3)°之间;Zn-O键长在0.201 8(5)~0.224 4(5)nm之间,Zn-N键长为0.205 0(6)~0.206 7(5)nm。在配合物1~3中噻吩甲酰腙的羰基均发生了烯醇化重排。而从图4b可见,在3中Baf-的呋喃甲酰腙片段依然保持着酰胺的结构,发生烯醇化转变的是苯甲酰基片段的羰基。这种情况与杜慷慨[11]报导的苯甲酰基丙酮缩水杨酰腙镍配合物中2个羰基都发生烯醇化的情况又不同。由于羰基烯醇化并去质子化,与Zn(Ⅱ)金属离子的静电引力增强,所以Zn1-O1和Zn1-O4的键长要比Zn1-O2和Zn1-O5短很多。酰腙配体烯醇化的双键C7=C8与亚胺基C9=N1以及次氨基N2与C9=N1和呋喃甲酰基的p-π共轭,形成一个离域共轭大平面,在大平面与苯环的二面角为44.5°;另一非对称配体也存在类似情况,二面角为28.5°。

图4 (a)配合物3前驱体Pzl的分子结构;(b)配合物3的分子结构Fig.4 (a)Molecular structure of precursor Pzl of 3;(b)Molecular structure of 3
2.2 红外光谱特征
配合物1~3的IR中在1 593~1 614 cm-1都出现了一个归属νC=N的强吸收峰,同时在931~987cm-1可以观察到νN-N的吸收峰;由于酰胺的羰基烯醇化,所以1~2中未出现来自次氨基-NH-的νNH,而在3中可以看到3 133 cm-1处νNH的尖锐的强吸收峰,其前驱体Pzl则未出现νNH,而是在3 409 cm-1处出现了一个来自羟基伸缩振动的强吸收峰。1中1 655 cm-1处的强吸收来自DMF的羰基伸缩振动,1 291 cm-1处极强的吸收归属于硝基-NO2的对称伸缩振动;1 424、854和730 cm-1的吸收峰则是phen的特征。3中同样出现了羰基的伸缩振动(1 628 cm-1),它来自呋喃酰基的片段,因为与金属离子配位,所以与前驱体Pzl(1 634 cm-1)相比向低波数方向位移。
2.3 紫外光谱特征
化合物DMF溶液的电子光谱见图5。图5a中配体H2Lss和配合物1中297、395 nm和295、401 nm的2对宽吸收带,对应于酰腙与芳环形成共轭体系的π→π*电子跃迁和分子内的电荷转移跃迁(ILCT)。1中268 nm吸收带来自phen的π→π*电子跃迁,310 nm可能与n→π*电子跃迁有关。配合物2(图5b)在264和351 nm处的吸收带归属芳环的π→π*电子跃迁和配体分子内的荷移跃迁,与配体H3Ldis(261、341 nm)相比略有红移。由图5b可见,配合物3与其前驱体相比电子跃迁行为有明显不同。Pzl在266和310 nm处的吸收可指认为苯环的π→π*电子跃迁和C=N与C=O的n→π*电子跃迁。与金属配位后Pzl开环转变为Baf-,Baf-的苯甲酰基烯醇化的双键与亚胺基以及次氨基与亚胺基和呋喃甲酰基的p-π共轭,因为形成了巨大的离域共轭体系,从而在406 nm出现了很强的分子内荷移跃迁吸收带,使n→π*电子跃迁强度减弱并红移。
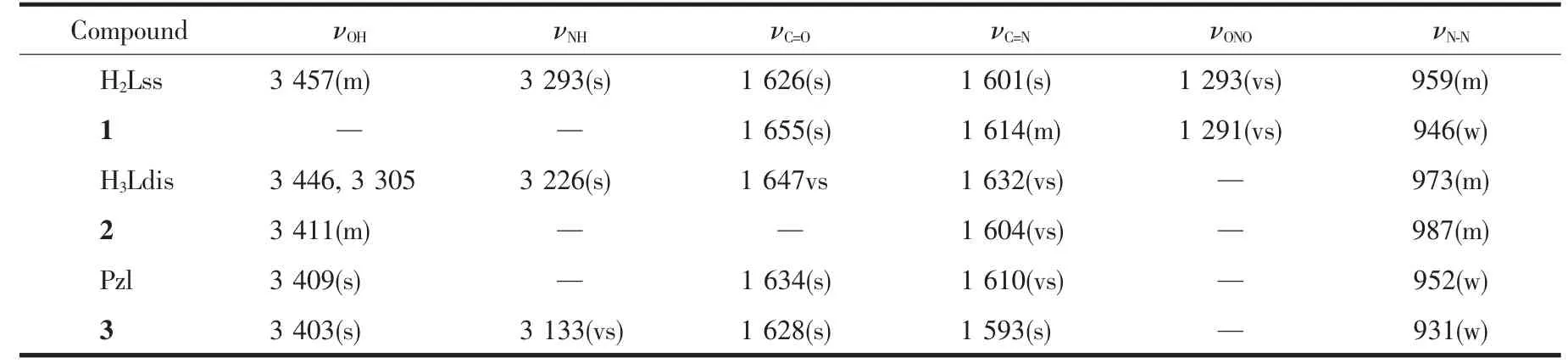
表3 化合物红外光谱的主要特征Table 3 Main characteristic peaks of infrared spectrum of the compounds cm-1
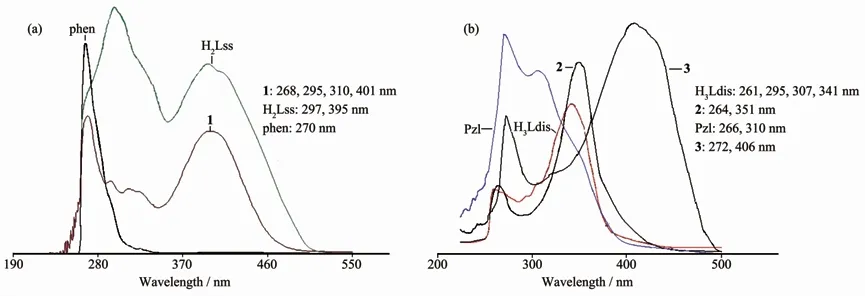
图5 化合物的紫外可见光谱Fig.5 UV-Vis spectra of the compounds

图6 配合物的热重分析图Fig.6 TG curves of the complexs
2.4 配合物的热稳定性
在N2气氛的保护下,升温速率10℃·min-1,配合物的热重分析见图6。3种配合物第一阶段的失重是因为失去溶剂分子DMF和甲醇所致,配合物1于85~176℃失重11.8%(理论值为12.02%),配合物2于室温至110℃失重8.8%(理论值为9.09%),配合物3于室温至112℃失重3.9%(理论值为5.04%)。第二阶段配合物骨架分解并快速挥发,分解温度分别为375℃(1)、364℃(2)和336℃(3),可见三者的热稳定性都较好。不同的是1和3在420℃之后仍在失重,至800℃时残重13%和17.5%;而2在522℃之后基本恒重,残重27.9%。
猜你喜欢
河南科技(2022年20期)2022-11-23
原子与分子物理学报(2022年3期)2022-03-05
世界农药(2021年3期)2021-12-09
辽宁科技大学学报(2021年2期)2021-07-22
中国食品(2020年9期)2020-05-26
表面技术(2020年2期)2020-03-04
青岛大学学报(工程技术版)(2019年2期)2019-09-10
分析化学(2017年6期)2017-06-15
山东工业技术(2016年15期)2016-12-01
中学化学(2015年8期)2015-12-29
