
本征空位缺陷对ZnO: Mn体系电子特性及磁性的影响
2019-12-06 09:31:52李俊贤符斯列王春安鲍佳怡丁罗城
原子与分子物理学报 2019年6期
李俊贤,符斯列,王春安,鲍佳怡,丁罗城,雷 涛
(1.华南师范大学物理与电信工程学院 广东省量子调控工程与材料重点实验室,广州 510006;2.华南师范大学华南先进光电子研究院,广州 510006; 3.广东技术师范学院电子与信息学院, 广州 510665)
1 引 言
ZnO是一种II-VI族宽禁带(Eg=3. 37 eV)直接带隙的化合物半导体材料,常温常压下的热力学稳定相是六方纤锌矿结构. 其室温下激子束缚能高达60 meV,高于GaN等半导体材料[1, 2]. 由于ZnO优异的光电性能,在发光二极管、光电探测器、表面声波器件及太阳能电池等领域有广泛的应用前景[3].
稀磁半导体(Diluted Magnetic Semiconductors,DMSs)是由磁性过渡金属元素或稀土金属元素部分取代半导体中的非磁性阳离子所形成的一类新型的半导体合金材料[4]. 近年来,Mn掺杂ZnO基稀磁半导体的电磁特性研究比较广泛. 相比于其他过渡金属元素掺ZnO体系,Mn具有力矩大、与Zn离子半径差较小,且具有半填充3d轨道的优点,有利于稳定掺杂[5]. 理论计算方面,2000年,Dietl等通过理论计算发现Mn掺杂的ZnO在室温将表现出铁磁行为,激发了人们对ZnO基稀磁半导体的研究兴趣[6];2013年Yao等人第一性原理计算了VO对Mn等过渡金属掺杂ZnO,发现VO会降低掺杂体系对红外光的吸收率[7];2019年,谢建明等人第一性原理计算了Mn掺杂ZnO纳米线,结果表明掺杂增加了纳米线的稳定性[8]. 在实验研究上,2001年Jin等人研究了制备ZnO薄膜中过渡金属固浓度的极限,证明Mn等过渡金属元素在ZnO中有很高的溶解度[9];2016年Gao等人共沉淀法制备出了Mn掺杂ZnO纳米粒子,并通过分析可见光致发光,发现氧空位随Mn浓度的增加而增加[10].
实验研究中用过渡金属元素Mn掺杂ZnO晶体很容易增加空位缺陷等本征点缺陷产生的几率,但是目前在本征空位缺陷对Mn掺ZnO电磁特性影响的理论计算方面,鲜有研究. 为了进一步了解空位缺陷对ZnO: Mn体系的影响,本文采用第一性原理计算方法分别研究了三种不同位置VZn和VO空位缺陷对ZnO: Mn体系电磁特性的影响,希望为实验研究提供有一定的理论参考.
2 模型构建与计算方法
2.1 模型构建
理想的ZnO是六方纤锌矿结构,由Zn的六角密排和O的六角密排沿c轴平移0.3825c相互位移套构而成,属于p63mc空间群,晶格常数a=b=3.249 Å,c=5.205 Å.α=β=90°,γ=120°. 其中c/a=1.602,稍小于理想六角密堆积结构的1.610[2].c轴方向上的Zn-O键长为1.991 Å,其他方向上的键长为1.973 Å[11]. 构建模型为2×2×2超晶胞,该超晶胞内有16个Zn原子,16个O原子,共计32个原子. 计算时用一个Mn代替一个Zn构成Mn掺杂,其掺杂浓度为6.25%. 空位则是除去超晶胞内部的一个Zn形成VZn,或者去掉一个O形成VO,其空位缺陷浓度为6.25%. 根据VZn、VO缺陷与Mn相对位置的远近分别构建了3种类型的ZnO: Mn空位缺陷体系模型,其模型如图1(a)-(f)所示. 其中ZnO: Mn-VZn体系分别为:ZnO: Mn-VZn1(近邻锌空位)、ZnO: Mn-VZn1(次近邻锌空位)、ZnO: Mn-VZn1(远近邻锌空位); ZnO: Mn-VO体系分别为:ZnO: Mn-VO1(近邻氧空位)、ZnO: Mn-VO2(次近邻氧空位)、ZnO: Mn-VO3(远近邻氧空位).
2.2 计算方法
计算软件采用基于密度泛函理论(Density Functional Theory,DFT)平面波赝势展开的CASTEP模块. 采用广义梯度近似(Generalized Gradient Approximation, GGA)修正的Perdew-Burke-Emzerhof (PBE) 泛函处理交换关联能,用超软赝势来描述价电子和离子实之间的相互作用. 几何优化的截断能Ecut=450 eV,第一布里渊区的k空间网格点为4×4×2. 几何优化参数设置如下:迭代过程中的收敛精度为 1.0×10-5eV·atom-1,原子间相互作用力收敛标准为0.03 eV·Å-1,最大位移为0.001 Å·atom-1,内应力不超过0.05 GPa. 价电子组态分别为:O:2s22p4,Zn:3d104s2,Mn:3d54s2.
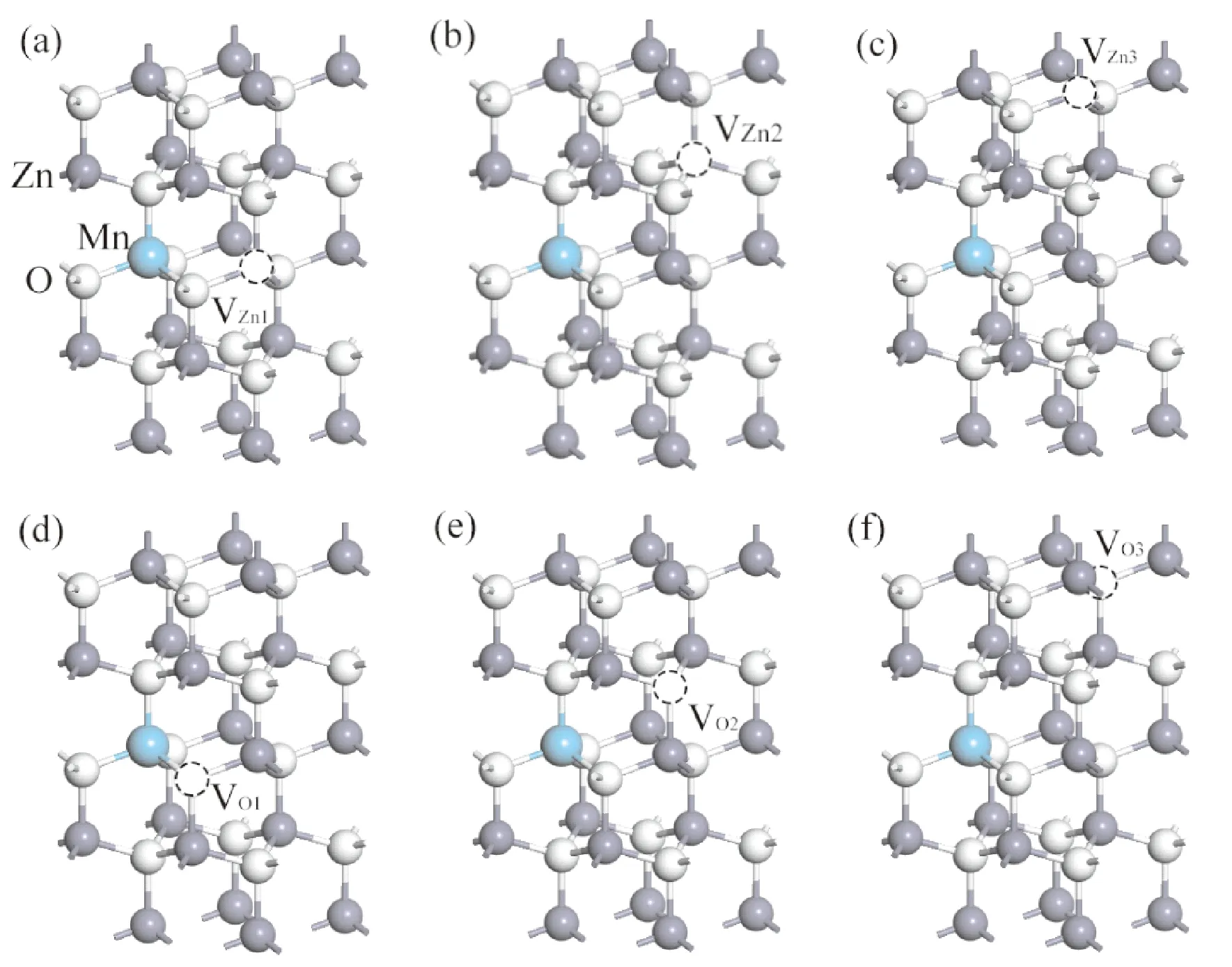
图1 超晶胞模型 (a)ZnO: Mn-VZn1;(b) ZnO: Mn-VZn2;(c) ZnO: Mn-VZn3; (d) ZnO: Mn-VO1;(e) ZnO:Mn-VO2; (f) ZnO: Mn-VO3Fig. 1 The supercell models (a) ZnO: Mn-VZn1;(b) ZnO: Mn-VZn2;(c) ZnO: Mn-VZn3; (d) ZnO: Mn-VO1;(e) ZnO: Mn-VO2;(f) ZnO: Mn-VO3
3 结果与讨论
3.1 结构与形成能分析
首先对所有掺杂体系进行晶格优化,优化后的晶格参数以及形成能如表1所示. 其中缺陷或者杂质X的形成能Ef[X]定义公式[12]为:
(1)
其中,Etot[X]是杂质或者缺陷体系的总能量;Etot[ZnO]是ZnO晶体的总能量;ni代表掺入或者移除原子的个数,掺入为正,移除为负;μi表示该原子的化学势,取为体系中平均到每个原子的基态能量.
从表1中得知,虽然Mn原子半径(0.83 Å)半径略大于Zn原子半径(0.74 Å)[13],但含空位缺陷的ZnO: Mn体系的体积仍略小于本征ZnO体系. ZnO: Mn-VZn体系的形成能要明显小于含ZnO: Mn -VO体系,说明ZnO: Mn体系中VZn相比于VO更容易形成. 其中ZnO:Mn-VZn1体系的形成能略小于其他两种ZnO: Mn-VZn体系,因此推测在ZnO: Mn体系中,VZn更容易在Mn最近邻位置产生;而通过比较三种ZnO: Mn-VO体系的形成能,同样得知VO更容易在Mn最近邻位置产生.
表1 各ZnO体系结构优化后的晶格参数以及缺陷形成能
Table 1 The lattice parameters and the defect formation energy of each ZnO system after geometry optimized

dopedZnOsystemsa(Å)C(Å)c/aV/(Å3)Ef(eV)ZnO3.2935.3011.610396.949—VZn3.2885.2941.61394.9157.074VO3.2705.3301.630387.3568.802ZnO:Mn3.3025.3181.610400.468-7.239ZnO:Mn-VZn13.3255.2611.582399.060-1.422ZnO:Mn-VZn23.3185.2501.582398.240-1.085ZnO:Mn-VZn33.0315.3581.768395.871-1.060ZnO:Mn-VO13.2715.2511.608389.3941.398ZnO:Mn-VO23.2805.2591.603390.8501.596ZnO:Mn-VO33.2815.2551.602390.8741.594
3.2 能带结构和态密度分析
图2(a)、(b)分别为本征ZnO的能带结构和分波态密度图,图2(a)只给出了-3.0-5.0 eV之间的能带结构以方便分析. 图2(a)中导带底和价带顶在同一G点,证实了本征ZnO为直接带隙半导体,带隙宽度Eg=0.731 eV,同文献[14], [15]一致,但远小于实验值(3.37 eV)[2],这是第一性原理计算普遍较低的结果,但是不影响我们对体系电子结构的理论分析[16]. 图2(b)中ZnO的价带分为上价带和下价带,上价带主要来自O 2p态,而下价带则是由Zn 3d贡献[17];导带部分主要来自Zn 4s态和O 2p态.
图2(c)、 (d)分别为Mn掺杂ZnO体系的能带结构和自旋分波态密度图. 其中,图2(c)表明ZnO: Mn体系仍为直接带隙半导体,且Mn的掺杂使费米能级向高能方向移动并进入导带,自旋向上部分在费米能级附近引入杂质能带[11]. 图2(d)在-2.0-0 eV内Mn 3d态电子的峰值与O 2p态重叠,表明Mn 3d态与O 2p态存在强烈的杂化作用,费米能级附近有很强的局域性,体系呈现半金属性[18].
计算得到各体系的能带结构和态密度如图3(a)-(f)所示. VZn在未掺杂的ZnO体系中是受主缺陷[19]. 而图3(a)、(c)、(e)均表明ZnO:Mn中VZn的出现使体系在价带顶引入了受主杂质能级,并且费米能级向低能区移动,因此VZn在ZnO:Mn体系中同样也是受主缺陷. 此外,在不同VZn位置下的ZnO: Mn体系中,随着VZn与Mn的距离越远,体系的费米能级向低能区的移动越微小. 但是三种体系的带隙宽度与无缺陷的ZnO:Mn体系相比没有明显改变,且仍为直接带隙. 同时,相比于图2(c) ZnO:Mn体系的态密度,图3(a)、(c)、(e)中ZnO: Mn-VZn体系的费米能级向价带低能区移动,使得费米能级处态密度增加,提高了体系的导电性. 而且随着VZn与Mn距离越远,费米能级处的态密度越大,导电性越强.
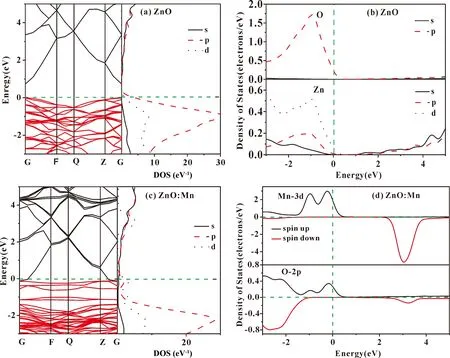
图2 (a)本征ZnO体系的能带结构和态密度;(b)本征ZnO体系的分波态密度; (c) ZnO: Mn体系的能带结构和态密度;(d) ZnO: Mn体系的自旋分波态密度Fig. 2 (a)The band structure and density of states of the intrinsic ZnO system; (b)the partial density of states of the intrinsic ZnO system; (c)the band structure and density of state of ZnO:Mn system; (d)the spin-polarized partial density of states of ZnO: Mn system

图3 ZnO: Mn 掺杂体系能带结构和态密度图 (a) ZnO: Mn-VZn1;(b) ZnO: Mn-VO1;(c) ZnO: Mn-VZn2;(d) ZnO: Mn-VO2;(e) ZnO: Mn-VZn3;(f) ZnO: Mn-VO3 Fig.3 The band structures and densities of states of ZnO: Mn systems: (a) ZnO: Mn-VZn1; (b) ZnO: Mn-VO1; (c) ZnO: Mn-VZn2; (d) ZnO: Mn-VO2; (e) ZnO: Mn-VZn3; (f) ZnO: Mn-VO3
VO在未掺杂的ZnO体系中是施主缺陷[20]. 而图3(b)、(d)、(f)均表明ZnO: Mn中VO的出现使体系在导带底引入了施主杂质能级,并且费米能级向高能区移动,因此VO在ZnO:Mn体系中同样也是施主缺陷. 另外,相比于无缺陷的ZnO: Mn,ZnO: Mn-VO体系禁带变宽十分明显,且变为间接带隙. 而且在不同VO位置的ZnO:Mn体系中,随着VO与Mn距离越远,带隙越窄,其中ZnO: Mn-VO1体系的带隙(Eg)为1.584 eV. 同时,相比于图2(c) ZnO:Mn体系的态密度,图3(b)、(d)、(f)中ZnO: Mn-VO体系的费米能级向高能方向移动,使得费米能级处态密度减少,降低了体系的导电性. 而且随着Mn与VO的位置越远,费米能级处态密度越大,所以导电性越强,但是低于ZnO: Mn体系.
3.3 磁性分析
图4(a)、(b)分别为ZnO: Mn-VZn和ZnO: Mn-VO体系的自旋分波态密度,表2为各体系中Mn、Zn、O原子的磁矩大小. 因为材料的磁性主要取决于费米面附近的电子分布,我们主要讨论费米能级附近的Mn 3d和O 2p态,其他态的电子贡献较小暂不考虑.
图4(a)中ZnO: Mn-VZn体系与图2(d)的ZnO: Mn相比Mn 3d与O 2p电子轨道杂化作用明显减弱,Mn 3d态自旋向上的电子态密度在费米能级处峰值明显降低,自旋劈裂现象减弱. 三种不同VZn位置下ZnO: Mn-VZn体系的费米能级附近Mn 3d自旋极化的峰值基本相同. 同时,表2中,相比于无缺陷的ZnO: Mn体系,VZn的引入导致ZnO: Mn体系总磁矩明显减少,体系的磁性减弱. 但是不同VZn位置下ZnO: Mn掺杂体系中总磁矩基本不变,与图4(a)的结果一致. 这说明VZn使ZnO: Mn体系的磁性减弱,且与VZn的位置无关.
图4(b)中ZnO: Mn-VO2、ZnO: Mn-VO3与图2(d)的ZnO:Mn相比Mn 3d与O 2p杂化作用没有减弱,自旋极化态密度峰峰值基本不变,而ZnO: Mn-VO1体系的自旋极化态密度峰值则有所降低. 但是表2的计算结果表明三种不同位置VO下ZnO: Mn体系的总磁矩同无缺陷的ZnO: Mn基本一致,都保持在5.0 μB左右. 这说明ZnO:Mn中无论VO在Mn的近邻、次近邻、远近邻位置产生,均不会对体系的磁性产生影响.
4 结 论
本文采用基于密度泛函理论的第一性原理赝势法,计算分析了ZnO: Mn掺杂体系中本征空位缺陷VZn和VO分别出现在相对Mn为近邻、次近邻、远近邻位置时体系的晶格结构、形成能、能带结构、态密度和磁矩. 结果表明:含空位缺陷的ZnO: Mn体系比本征ZnO的体积略小,其中在ZnO: Mn体系中VZn比VO更易产生,且两种缺陷更容易在Mn的近邻位置产生. 另外,相比于ZnO: Mn体系,ZnO: Mn-VZn体系的带隙没有明显改变,仍为直接带隙,体系电导率提高,并且随着VZn与Mn的距离变远,导电性增强. 但是VZn的存在会明显降低体系铁磁性,且与VZn的位置无关. 相比于ZnO: Mn体系,ZnO: Mn-VO体系的禁带宽度变大,且变为间接带隙,体系的导电率下降,并且随着VO与Mn距离变远,导电性上升,但仍小于ZnO: Mn体系. 此外,无论与Mn的距离远近,VO的引入均不会影响ZnO: Mn体系的铁磁性.
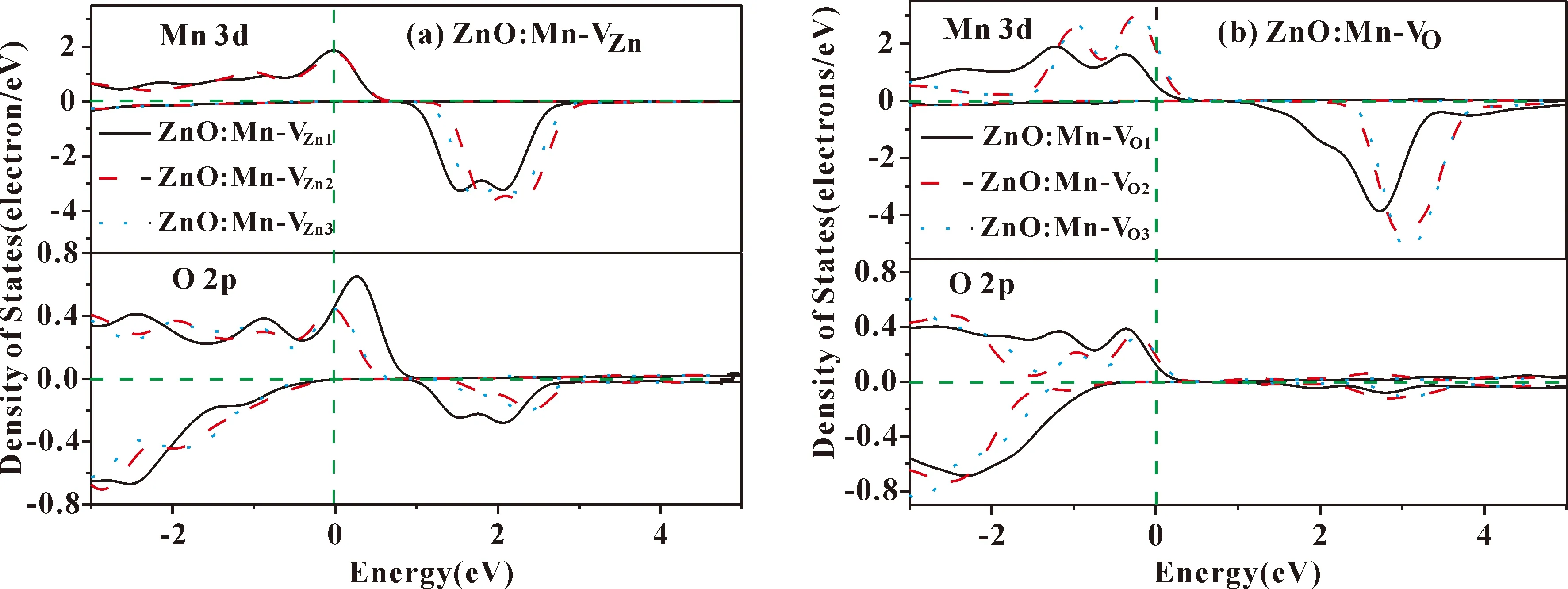
图4 ZnO: Mn掺杂体系的自旋分波态密度 (a)ZnO: Mn-VZn;(b) ZnO: Mn-VOFig. 4 The spin-polarized partial densities of states of ZnO: Mn systems: (a) ZnO: Mn-VZn; (b)ZnO: Mn-VO
表2 ZnO: Mn掺杂体系中各原子的磁矩
Table 2 The magnetic moment of each atom of ZnO:Mn doping systems

dopedZnOsystemsMn磁矩/μBZn磁矩/μBO磁矩/μB总磁矩/μBZnO:Mn4.730.030.234.99ZnO:Mn-VZn13.240.08-0.323.00ZnO:Mn-VZn23.640.08-0.732.99ZnO:Mn-VZn33.540.10-0.643.00ZnO:Mn-VO14.80-0.090.295.00ZnO:Mn-VO24.75-0.020.275.00ZnO:Mn-VO34.750.010.245.00
猜你喜欢
吉首大学学报(自然科学版)(2023年6期)2023-12-22 08:18:20
农业工程学报(2022年7期)2022-07-09 07:06:56
数学物理学报(2020年1期)2020-04-21 06:00:22
数学物理学报(2018年4期)2018-09-14 03:40:58
吉首大学学报(自然科学版)(2018年3期)2018-07-03 03:14:12
Chinese Journal of Chemical Engineering(2017年5期)2017-05-28 10:22:54
深圳大学学报(理工版)(2015年6期)2015-11-26 12:33:48
河南科技(2014年23期)2014-02-27 14:18:52
物理通报(2011年9期)2011-01-24 07:39:36
物理学报(2010年6期)2010-09-08 06:05:24
