
Dubin-Johnson综合征分子遗传学研究进展
2019-12-06 05:56张红艳李亚绒张艳敏车凤玉
中国妇幼健康研究 2019年11期
张红艳,李亚绒,张艳敏,车凤玉
(1.西安医学院,陕西 西安 710021;2.西安市儿童医院,陕西 西安 710003)
Dubin-Johnson综合征(Dubin-Johnson syndrome,DJS)由Dubin和Johnson于1954年首次发现并报道[1]。DJS是一种罕见的常染色体隐性遗传病,目前报道西帕迪奇犹太人群中发病率较高,约1∶3 000[2]。ATP结合盒C亚家族成员ABCC2基因突变致多药耐药相关蛋白(multidrug resistance associated proteins,MRP2)功能障碍或缺失是DJS发病的重要机制。临床以持续性或间歇性皮肤、巩膜及小便发黄(轻至中度)为主要表现,可有右上腹不适或隐痛、乏力、食欲不振、恶心、呕吐等表现,约半数患者可有轻度肝脏肿大或轻微肝区压痛,脾肿大者少见,无皮肤瘙痒[3]。DJS患者肝脏外观呈黑色,组织学表现为肝小叶中央区肝细胞溶酶体内有棕褐色色素颗粒沉着,肝脏病理活检为确诊的金准标。DJS临床症状一般比较轻微,预后良好,一旦确诊无需特殊治疗,口服熊去氧胆酸和苯巴比妥钠对症即可[1]。本文就DJS分子遗传学研究进展进行综述。
1 MRP2的结构
MRP2是一种190kDa的完整膜糖蛋白,含有1 545个氨基酸,它包含两个ATP结合磁带和17个跨膜序列,主要分布于肝细胞的极化上皮根尖小管膜上,以及其他具有极化细胞的顶膜上,如肠细胞和肾小管细胞,是一种ATP依赖的两亲阴离子输出泵,可用于共轭和非共轭两亲阴离子的输出。MRP2的共轭输出泵及其特异性小管异构体,使其底物很广泛,包括非胆汁酸有机阴离子转运体、多特异性有机阴离子转运体、谷胱甘肽S共轭输出泵及白三烯输出泵[4-5]。MRP2功能障碍或缺失,使得肝细胞中结合胆红素及其他有机阴离子向毛细胆管排泄障碍,引起胆红素在体内淤积,从而导致慢性的、以结合胆红素升高为主的高胆红素血症[6]。
2 MRP2的编码基因ABCC2及其突变
MRP2的编码基因ABCC2(ATP结合盒C亚家族成员)于1997年被Paulusma等在高胆红素血症大鼠中发现,同年谷口一等在顺铂耐药肿瘤细胞中发现了人类ABCC2基因,该基因位于10号染色体短臂24带(10q24),1999年Tsujii等[7]发现该基因由32个外显子组成。迄今为止已报导的DJS相关的ABCC2基因突变位点多达68个,包括:错义突变、无义突变、剪接突变、调节突变、删除/缺失突变、插入突变、小插入-缺失突变、复杂重排突变,其中绝大多数是碱基置换突变导致的错义突变和无义突变,见表1。

表1 Dubin-Johnson综合征编码基因ABCC2突变类型
续上表
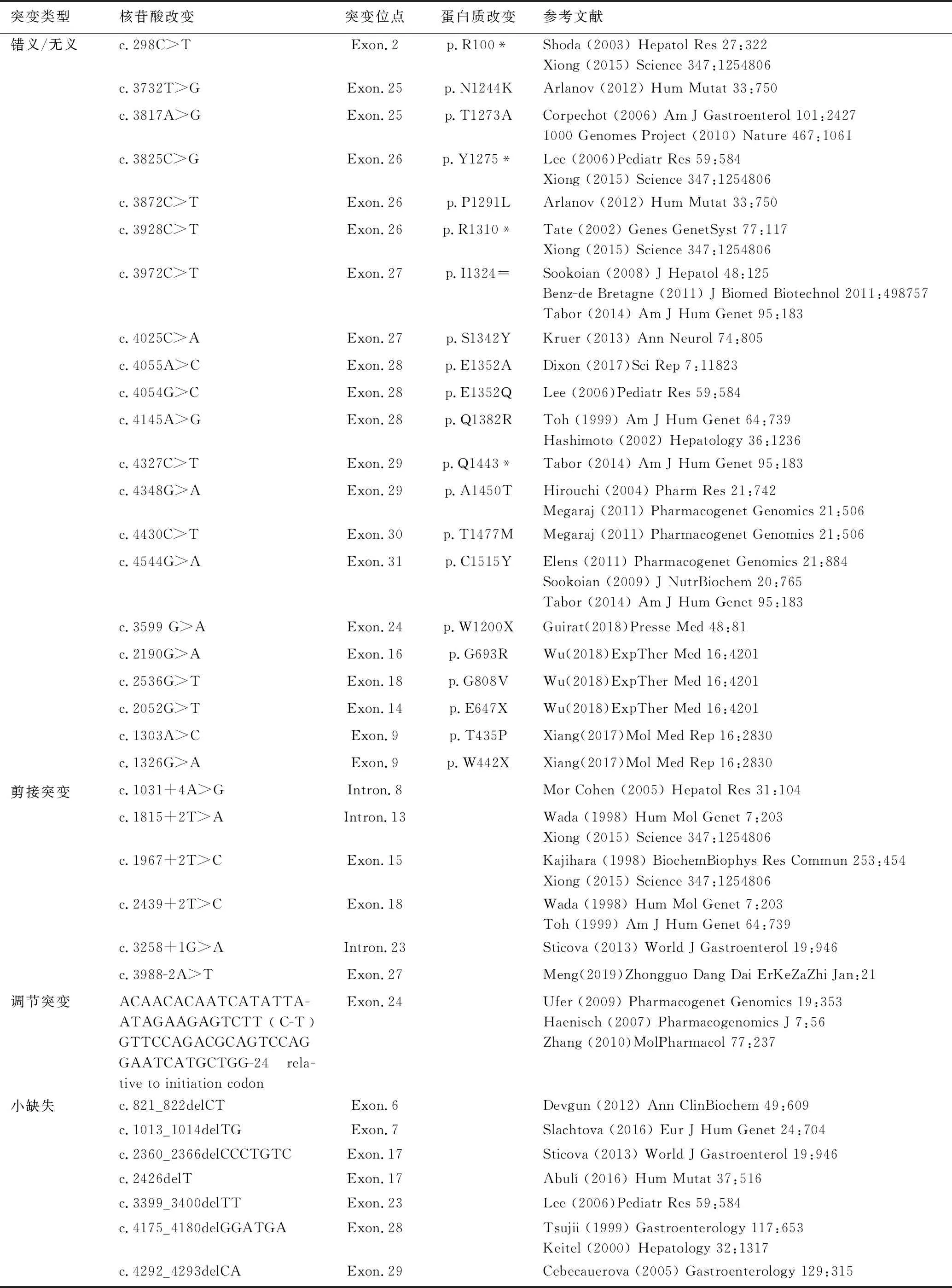
突变类型核苷酸改变突变位点蛋白质改变参考文献错义/无义c.298C>TExon.2p.R100*Shoda(2003)HepatolRes27:322Xiong(2015)Science347:1254806c.3732T>GExon.25p.N1244KArlanov(2012)HumMutat33:750c.3817A>GExon.25p.T1273ACorpechot(2006)AmJGastroenterol101:24271000GenomesProject(2010)Nature467:1061c.3825C>GExon.26p.Y1275*Lee(2006)PediatrRes59:584Xiong(2015)Science347:1254806c.3872C>TExon.26p.P1291LArlanov(2012)HumMutat33:750c.3928C>TExon.26p.R1310*Tate(2002)GenesGenetSyst77:117Xiong(2015)Science347:1254806c.3972C>TExon.27p.I1324=Sookoian(2008)JHepatol48:125Benz-deBretagne(2011)JBiomedBiotechnol2011:498757Tabor(2014)AmJHumGenet95:183c.4025C>AExon.27p.S1342YKruer(2013)AnnNeurol74:805c.4055A>CExon.28p.E1352ADixon(2017)SciRep7:11823c.4054G>CExon.28p.E1352QLee(2006)PediatrRes59:584c.4145A>GExon.28p.Q1382RToh(1999)AmJHumGenet64:739Hashimoto(2002)Hepatology36:1236c.4327C>TExon.29p.Q1443*Tabor(2014)AmJHumGenet95:183c.4348G>AExon.29p.A1450THirouchi(2004)PharmRes21:742Megaraj(2011)PharmacogenetGenomics21:506c.4430C>TExon.30p.T1477MMegaraj(2011)PharmacogenetGenomics21:506c.4544G>AExon.31p.C1515YElens(2011)PharmacogenetGenomics21:884Sookoian(2009)JNutrBiochem20:765Tabor(2014)AmJHumGenet95:183c.3599G>AExon.24p.W1200XGuirat(2018)PresseMed48:81c.2190G>AExon.16p.G693RWu(2018)ExpTherMed16:4201c.2536G>TExon.18p.G808VWu(2018)ExpTherMed16:4201c.2052G>TExon.14p.E647XWu(2018)ExpTherMed16:4201c.1303A>CExon.9p.T435PXiang(2017)MolMedRep16:2830c.1326G>AExon.9p.W442XXiang(2017)MolMedRep16:2830剪接突变c.1031+4A>GIntron.8MorCohen(2005)HepatolRes31:104c.1815+2T>AIntron.13Wada(1998)HumMolGenet7:203Xiong(2015)Science347:1254806c.1967+2T>CExon.15Kajihara(1998)BiochemBiophysResCommun253:454Xiong(2015)Science347:1254806c.2439+2T>CExon.18Wada(1998)HumMolGenet7:203Toh(1999)AmJHumGenet64:739c.3258+1G>AIntron.23Sticova(2013)WorldJGastroenterol19:946c.3988-2A>TExon.27Meng(2019)ZhongguoDangDaiErKeZaZhiJan:21调节突变ACAACACAATCATATTA-ATAGAAGAGTCTT(C-T)GTTCCAGACGCAGTCCAGGAATCATGCTGG-24rela-tivetoinitiationcodonExon.24Ufer(2009)PharmacogenetGenomics19:353Haenisch(2007)PharmacogenomicsJ7:56Zhang(2010)MolPharmacol77:237小缺失c.821_822delCTExon.6Devgun(2012)AnnClinBiochem49:609c.1013_1014delTGExon.7Slachtova(2016)EurJHumGenet24:704c.2360_2366delCCCTGTCExon.17Sticova(2013)WorldJGastroenterol19:946c.2426delTExon.17Abulí(2016)HumMutat37:516c.3399_3400delTTExon.23Lee(2006)PediatrRes59:584c.4175_4180delGGATGAExon.28Tsujii(1999)Gastroenterology117:653Keitel(2000)Hepatology32:1317c.4292_4293delCAExon.29Cebecauerova(2005)Gastroenterology129:315
续上表
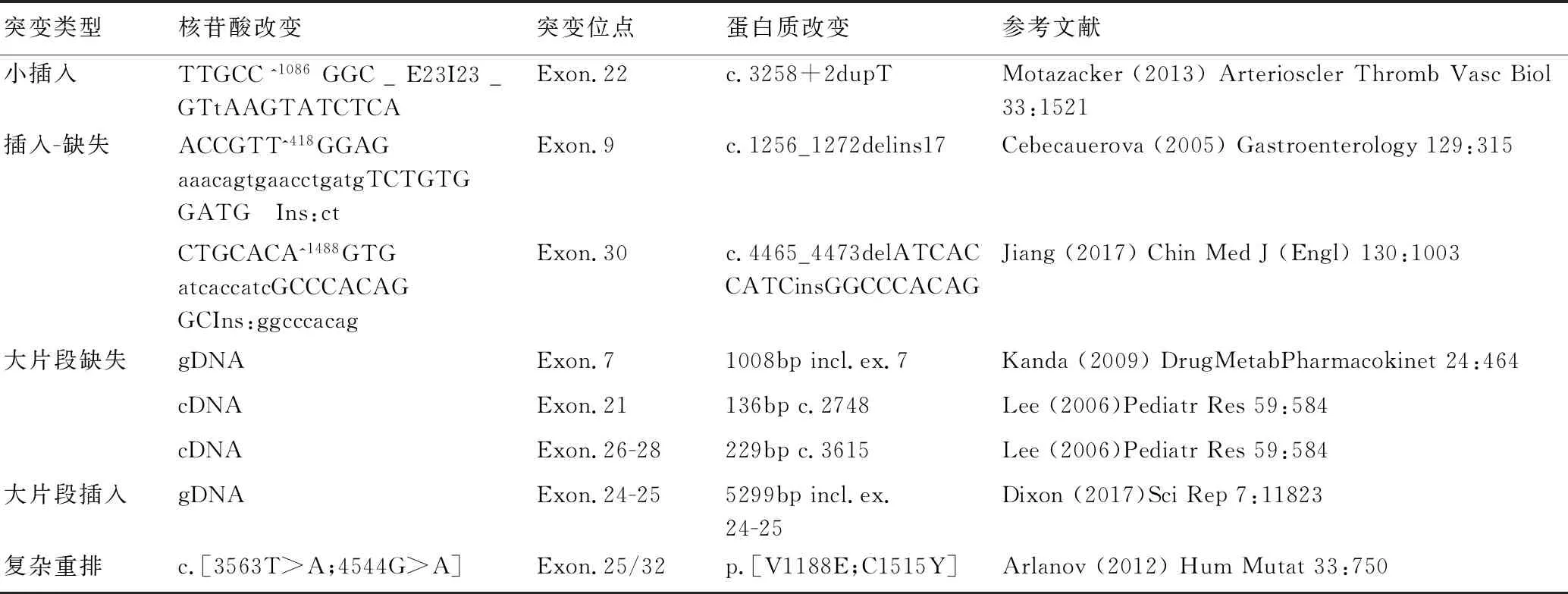
突变类型核苷酸改变突变位点蛋白质改变参考文献小插入TTGCC^1086GGC_E23I23_GTtAAGTATCTCAExon.22c.3258+2dupTMotazacker(2013)ArteriosclerThrombVascBiol33:1521插入-缺失ACCGTT^418GGAGaaacagtgaacctgatgTCTGTGGATG Ins:ctExon.9c.1256_1272delins17Cebecauerova(2005)Gastroenterology129:315CTGCACA^1488GTGatcaccatcGCCCACAGGCIns:ggcccacagExon.30c.4465_4473delATCACCATCinsGGCCCACAGJiang(2017)ChinMedJ(Engl)130:1003大片段缺失gDNAExon.71008bpincl.ex.7Kanda(2009)DrugMetabPharmacokinet24:464cDNAExon.21136bpc.2748Lee(2006)PediatrRes59:584cDNAExon.26-28229bpc.3615Lee(2006)PediatrRes59:584大片段插入gDNAExon.24-255299bpincl.ex.24-25Dixon(2017)SciRep7:11823复杂重排c.[3563T>A;4544G>A]Exon.25/32p.[V1188E;C1515Y]Arlanov(2012)HumMutat33:750
注:*代表翻译提前终止;=代表氨基酸变化相同。
2.1错义突变和无义突变
2018年Togawa等[8]招募了10名新生儿期出现胆汁淤积,最终诊断为DJS的患儿,其中9例日本患儿,1例中国患儿。8例患儿在新生儿期进行了肝组织穿刺活检术,3例为黑肝,6例经病理免疫组化研究证实缺乏MRP2表达。所有患儿均进行基因检测,共发现ABCC2基因11个致病性突变位点,其中c.2882 C>T,p.K961R为新的致病性突变,认为错义突变和无义突变共同阻止了MRP2在肝小管膜中的表达,从而可能导致DJS。
2017年Xiang等[9]对1例临床诊断DJS的患者进行基因测序,在ABCC2基因的第10外显子发现了2个新突变,即错义突变:c.1303A>C,p.T435P,遗传自母亲,无义突变c.1326G>A,p.W442X,遗传自父亲,符合DJS常染色体隐性遗传。对突变后MRP2蛋白质功能研究,证实了错义突变c.1303A>C,p.T435P和无义突变c.1326G>A,p.W442X破坏了MRP2蛋白质的功能,从而导致DJS,由此证实了该患者DJS的临床诊断。
2019年Guirat等[10]对1名表现为黄疸的9岁男童进行ABCC2基因测序,在第25外显子发现一个新的致病突变,即无义突变:c.3599 G>A,p.W1200X,为纯合突变,遗传来自父母,从而诊断该患儿为DJS。本例患儿未进行肝组织穿刺活检。此外2014年Tabor等发现:c.3305 G>A,p.W1102*等新的致病性突变陆续被报道,至今已报道的错义突变和无义突变共有42个。
2.2剪接突变
2013年Sticova等[11]对一名癌症患者的随访中发现该患者表现为持续显著的高胆红素血症,肝组织穿刺活检、病理免疫组化研究证实了MRP2表达正常,基因测序确定了ABCC2基因的两个新致病性突变:即缺失突变:c.2360_2366delCCCTGTC和剪接突变:c.3258 + 1G>A,前者位于第18外显子,为杂合缺失;后者位于第23内含子,该突变在第23个剪接供体处发生了G>A的替换,上述位点的突变改变了RNA前体正常的剪接方式,引起mRNA的异常剪接,由此确诊该患者为DJS。2019年Meng等[12]通过基因检测确诊了1例DJS患儿,发现了ABCC2基因1个新的致病性突变:c.3988-2A>T,遗传自母亲。此类突变最早由Kajihara在1998年发现:c.1967+2T>C,随后相继发现 c.1815+2T>A,c.2439+2T>C和 c.1031+4A>G共6个剪接突变。
2.3缺失/删除突变
1999年Tsujii等[7]对2例DJS患者进行研究,检测了ABCC2基因32个外显子,以及外显子-内含子的边界区域,除了发现已报道的无义突变:c.1066 C>T,同时在2例患者发现了第1 392到1 394核苷酸之间6个核苷酸的缺失突变,致使其下游的开放阅读框架发生了改变,最终导致精氨酸和蛋氨酸的缺失,患者的肝细胞小管膜MRP2不表达。
2016年Slachtova等[13]对7个罗姆人家系56名成员进行研究,共发现17例DJS患者和30例杂合子携带者。该研究在ABCC2基因的第8外显子发现了一个新的缺失突变c.1013_1014 delTG,17例DJS患者均为纯合子的c.1013_1014 delTG,导致被编码的MRP2发生移码和截短,由原来的1545个氨基酸缩短为352个氨基酸。研究认为ABCC2基因的c.1013_1014 delTG突变可能导致MRP2的mRNA快速降解,从而导致MRP2的合成、稳定性及转运方面的缺陷。研究在7个无相关性的罗姆人家系中发现相同的c.1013_1014 delTG变异,推断c.1013_1014 delTG变异为罗姆人的热点变异。2016年Slachtova等[13]发现的ABCC2基因新的致病性缺失突变:c.2360_2366delCCCTGTC,该突变位于第18外显子,为第2 360到2 366核苷酸之间7个核苷酸的缺失突变,使其下游的开放阅读框架发生了改变,最终导致氨基酸的缺失,从而影响MRP2的功能。
2006年Lee等[14]对4例肝组织病理符合国内DJS诊断标准的患者进行研究,其中2名患者新生儿期即出现胆汁淤积并诊断,另外2例有新生儿黄疸和高胆红素血症史,于青春期诊断DJS,并对4例患者进行长达5~20年随访。在4例患者中共发现ABCC2基因的6个突变位点,其中5个突变位点为已报道的,包括缺失突变2748del136和3615del22,错义突变p.L441M和p.E1352Q以及无义突变p.Y1275*,此外还发现一个未经报道的杂合子的缺失突变:c.3399_3400delTT,该突变使得42个氨基酸移位及终止密码子提前出现,致使蛋白质合成中断,最终导致ABC结构域的丢失。该研究表明,MRP2中具有重要功能的ABC结构域的丢失可能与DJS的早期发病相关。
2009年Kanda等[15]对1例临床诊断为DJS患者基因测序,发现了ABCC2基因一个新的突变位点:gDNA 1008 bpincl.Ex.7,该突变为纯合的1008 bp的外显子7大片段缺失,患者未进行肝组织穿刺活检,无法确定外显子7的1 008 bp大规模缺失对MRP2功能的影响,推测该缺失影响MRP2合成。
迄今为止共发现的缺失/删除突变14个,其中11个小缺失、3个大片段缺失。
2.4小插入-缺失突变
2005年Cebecauerova等[16]报道一例3岁男孩携带ABCC2基因2个新的突变:其中一个为外显子10的杂合插入-缺失:indel 1256 insCT/delAAACAGTGAACCTGATG,遗传自父亲,另一个为外显子30的杂合缺失4292delCA,遗传自母亲,后者的突变导致了氨基酸的移位和1 461位提前出现了终止密码子。但患者肝脏并不表现为黑肝,且组织学正常,由此得出结论:对于诊断不明确的、怀疑有遗传性高胆红素血症的患者,无论其表型如何,无创的DNA测序分析应为首选方法,当基因测序不能明确时,才应选取有创的肝组织穿刺活检检查。
2017年Jiang等[17]对一名血清胆红素持续轻度升高的女性患者进行研究,发现了ABCC2基因新的突变c.4712_4720delinsGGCCCACAG(父源)和p.I448V(母源),两者均被证实是致病性突变,上述突变通过降低蛋白质的稳定性,严重破坏转运蛋白的活性,从而可能导致DJS。
2.5插入突变
2017年Dixon等[18]在对妊娠肝内胆汁淤积症患者的研究中,发现了ABCC2基因的一个新突变:gDNA Dup 5299 bp incl.ex.24-25,该突变是包括外显子24和25在内的5 299个碱基对的基因组重复,该研究预测这个突变会导致一个早熟的终止密码子提前插入,使得其下游的阅读框发生改变,使多肽链的氨基酸种类和序列发生改变。同时还发现一个已报道的错义突变,c.4055 A>C,可能导致p.Glu1352Ala的改变,为有害突变。迄今为止发现此类突变有2个,其中小插入1个、大片段插入1个。
3总结
ABCC 2基因的分子遗传学分析对DJS的诊断具有重要意义。通过基因检测可明确患者是否患有DJS,从而减少患者反复不必要的就诊,减少不必要的检查及治疗,同时还可减少肝组织穿刺活检等有创侵入性操作。近年来通过基因检测手段确诊的DJS患者逐年增加,新的致病性突变不断被发现,丰富了人类基因组突变数据库,使得人们对DJS的认识也更加全面。
猜你喜欢
安徽农业大学学报(2022年2期)2022-11-09
电子科技大学学报(2022年5期)2022-10-29
广西医科大学学报(2022年5期)2022-06-07
中国循证儿科杂志(2022年2期)2022-05-26
昆明医科大学学报(2022年3期)2022-04-19
中国土壤与肥料(2021年5期)2021-12-02
中国生殖健康(2020年4期)2021-01-18
中南医学科学杂志(2019年6期)2019-12-05
肿瘤预防与治疗(2019年6期)2019-07-30
中国动物保健(2015年4期)2015-10-21
